
Review Article/ Open Access
DOI: 2021; 3(2): 187 -199. doi: 10.31488/EJRM.115
A Review of Alkaline Phosphatase in Preventing Systemic Inflammation after Cardiac Surgery
Tony Vu1,Tyson A Fricke2,Julian A Smith*2,3
1. School of Medicine, Faculty of Medicine, Nursing and Health Sciences, Monash University, Melbourne, Australia
2. Department of Cardiothoracic Surgery, Monash Health, Melbourne, Australia
3. Department of Surgery (School of Clinical Sciences at Monash Health), Monash University, Melbourne, Australia
*Corresponding author: Professor Julian A. Smith, Head, Department of Surgery (School of Clinical Sciences at Monash Health), Monash University, Australia
Abstract
Globally, there has been an increasing number of cardiac surgery procedures performed year on year. The cardiac surgery patient population is becoming older and are more likely to possess numerous comorbidities such as diabetes and hypertension, thereby reducing their physiological reserve. Cardiac surgery, particularly on cardiopulmonary bypass, can induce a systematic inflammatory response due to surgical trauma, the interaction of blood with foreign surfaces in extracorporeal circulation, endotoxemia and ischemic reperfusion injury. The predominant molecular mediators of the systematic inflammatory response following cardiac surgery include the complement system, the coagulation cascade, cytokines, neutrophils, and the vascular endothelium. If the systemic inflammatory response is excessive, organ dysfunction may ensue. The organs commonly affected from the systemic inflammatory response following cardiac surgery include the heart, lungs, kidneys, brain, intestinal and the vascular endothelium. Alkaline phosphatase is a highly conserved metalloproteinase with four distinct isoforms in humans, including tissue non-specific, intestinal, placental and germ cell alkaline phosphatase. Alkaline phosphatase may dephosphorylate and reduce the toxicity of endotoxins, lipopolysaccharides, and extracellular nucleotides. In both animal studies and clinical trials, alkaline phosphatase appears to be a promising therapeutic agent, capable of attenuating the systemic inflammatory response to cardiac surgery.
Key words: Cardiac surgery, cardiothoracic surgery, cardiopulmonary bypass, inflammation, systemic inflammatory response syndrome, alkaline phosphatase
Introduction to Cardiac Surgery
Cardiothoracic surgery is a rapidly evolving specialty of medicine concerned with the management of multiple pathologies of organs within the thoracic cavity. At least two million cardiac surgeries are performed globally per annum, and this number continues to rise [1]. With an aging population, patients undergoing surgery are more likely to be older and possess more comorbidities such as diabetes and hypertension [2]. Patients undergoing cardiac surgery with cardiopulmonary bypass (CPB) experience systemic inflammation, resulting in post-operative complications and an increased risk of morbidity and mortality. Hence, therapeutic measures which limit the inflammatory response to cardiac surgery will improve surgical outcomes. This article will describe the inflammatory response which occurs following cardiac surgery performed on CPB and will review the use of alkaline phosphatase as a novel therapeutic measure to attenuate such.
The Spectrum of Inflammation
Inflammation is a normal, physiological response to tissue injury and insult [3]. This section will review the spectrum of inflammation, the molecular mediators of inflammation and describe how the use of CPB is associated with inflammation.
The systemic inflammatory response syndrome (SIRS) describes a non-specific inflammatory cascade independent of the cause. It is often considered as an extension of the physiological immune response [4]. The clinical diagnosis of SIRs requires 2 or more of the following criteria as listed in Table 1. Cardiac surgery is a well-established cause of SIRS. However, the proportion of patients which develop a florid SIRS following cardiac surgery is variable. All patients in Michalopoulos’s 1996 study developed SIRS during the first 12 post-operative hours. In contrast, Squiccimarro et al. 2019 stated that 28.3% of their cohort developed SIRS after 24 hours, while Boehne et al. 2017 had approximately 33% of their cohort develop SIRS [5, 6]. When the inflammatory response is tightly controlled by homeostatic and anti-inflammatory mechanisms, inflammation is generally self-limiting. Postulated by Roger Bone, an anti-inflammatory mechanism exists during sepsis, known as the compensatory or counter anti-inflammatory response (CAR). The balance between SIRS and CAR is critical in determining the prognosis of a surgical patient, with excessive inflammation associated with an increased incidence of post-operative complications and multi-organ dysfunction [7, 8]. Mortality following uncontrolled SIRS has been reported to be as high as 90% in various studies [9].
Table 1.Criteria for Diagnosis of Systemic Response Syndrome [4, 17, 18]
| Parameter | Value |
|---|---|
| Temperature | >38°C or <36 °C |
| Respiratory Rate | >20 breaths per minute or PaCO2 <32 mmHg |
| Heart Rate | >90 Beats per minute |
| White Blood Cell Count | >12,000 /L (Leucocytosis), <4000/L (Leukopenia) or >10% immature cells (i.e left shift) |
Recently, sepsis has been defined as life threatening organ dysfunction caused by a dysregulated host response to infection (Sepsis-3). Organ dysfunction is defined as an increase in 2 or more points in the sequential organ failure assessment (SOFA). The most common cause of sepsis is gram negative bacterial infection [10]. Sepsis is associated with an increase in mortality of greater than 10% and is [11, 12]. Sepsis is a significant contributor to morbidity and mortality, with a global incidence of 48.9 million cases, and accounts for 19.7% of global deaths in 2017. Health care spending on managing sepsis is significant, costing the United States government more than $20 billion USD per annum [13]. It is also paramount to prevent the development of sepsis, given that the survivors of sepsis are likely to experience long term physical or psychological sequalae [14, 15].
Septic shock is defined as a form of sepsis which may be clinically identified by the requirements for vasopressors to maintain a mean arterial pressure (MAP) >65 mmHg and serum lactate greater than 2mmol/L despite aggressive fluid resuscitation. Septic shock is associated with a mortality rate of greater than 40% [11, 16].
The Activators of the Inflammatory Response in Cardiac Surgery
During cardiac surgery with CPB, the inflammatory response is activated by numerous mechanisms including surgical trauma, the interaction of blood with the extracorporeal circulation, endotoxemia and ischemic reperfusion injury (Figure 1) [9,19].
Surgical trauma
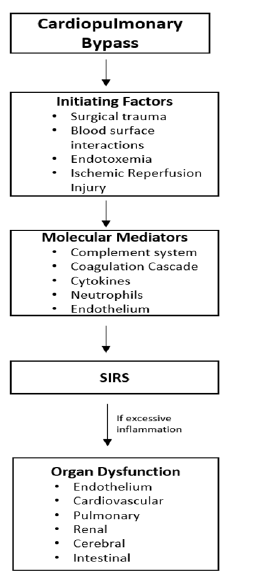
Figure1:A pathway depicting how cardiac surgery with cardiopulmonary bypass activates inflammatory mediators leading to a systemic inflammatory response syndrome (SIRS) and potentially organ dysfunction. Adapted from Laffey et al. 2002 (4)
The act of performing an incision, such as with a midline sternotomy in cardiac surgery, is associated with significant inflammation [19]. Trauma activates a significant immunological response to destroy microbial organisms and promote tissue healing [20]. Nevertheless, tissue destruction may also release toxic mediators into systemic circulation, exacerbating the inflammatory response [21].
Blood surface interactions during extracorporeal circulation
Contact activation of the immunological system occurs following an interaction between the patient’s blood with the foreign surfaces of the CPB circuitry [4]. This may result in thrombocytopenia, the release of platelet granular contents and the synthesis of thromboxane [22]. These changes may clinically manifest as haematological complications, particularly a prolonged bleeding time or potentially disseminated intravascular coagulation (DIC). Additionally, initiation of the complement cascade may also occur following contact activation [23].
Translocation of endotoxins from the bowel (Endotoxemia)
Endotoxins, also known as lipopolysaccharides (LPS), are pyrogens found within the cell wall of gram-negative bacteria. Structurally, they consist of a lipid A moiety, an inner core, an outer core and an O antigen. The lipid A moiety consists of two phosphorylated glucosamine saccharides, with the phosphates essential for toxicity [19]. Normally, endotoxins are produced by commensal bacteria located within the GIT and are confined to this location via endothelial cells. An interaction between endotoxins and immunological mediators such as TLR4 would promote an inflammatory response [24].
Endotoxins are released into systemic circulation during cardiac surgery with CPB. It was initially believed that the endotoxins found within the body could be contaminants derived from the environment, given they were also found in the pulmonary artery and CPB equipment (cardiac suction lines and arterial outlet oxygenators) [25]. However, it has been reported that environmental endotoxins are ubiquitous, and are less likely to be physiologically deleterious [26]. Currently, it is accepted that ischemic-reperfusion injury of the gastrointestinal tract results in increased permeability and facilities the translocation of endotoxins into the portal circulation. Excessive systemic endotoxin levels have been linked to organ dysfunction and multiorgan failure [27-29]. The duration of aortic cross clamping during CPB is also associated with the level of endotoxemia [19].
Ischemic reperfusion injury
Ischemic reperfusion injury is a phenomenon which involves tissue and organ damage following the restoration of blood flow to a previously ischemic region [30]. This may occur during cardiac surgery following the release of the aortic cross clamp. Aortic cross clamping is used during surgery to facilitate procedures on the heart under cardioplegic arrest [31]. The release of the aortic cross clamp is associated with the subsequent development of multi-organ dysfunction following reperfusion of the heart and other organs [32].
There are numerous causes of reperfusion injury. Firstly, the production of radical oxygen species (ROS) may occur following reperfusion of the previously ischemic organ [33]. ROS may alternatively be generated via the activity of xanthine oxidase and through neutrophil activation and migration [34]. Superoxide anion and peroxynitrite are examples of ROS which appear to be the predominant mediators during the initial phase of ischemic reperfusion injury [35]. Additionally, ischemia promotes the intracellular accumulation of hydrogen ions, calcium ions and other cellular toxins resulting in acidosis. With reperfusion, there is a rapid alteration in ionic movement to correct the acidosis. However, this paradoxically results in cellular cytotoxicity [36]. Moreover, the accumulation of intracellular calcium ions coupled with the enhanced entry of calcium ions due to ionic corrections during reperfusion result in enhanced myofibrillar contractility and myocardial stunning [34]. Myocardial stunning refers to a prolonged, yet reversible dysfunction of the myocardium following reperfusion injury [33, 37].
The Underlying Molecular Mediators of Inflammation
There are several molecular mediators of inflammation during cardiac surgery, including the complement system, the coagulation cascade, cytokines, neutrophils, nitric oxide and the endothelium.
The complement system
The complement system consists of over 30 serum proteases which are systematically cleaved to produce active proteins responsible for opsonisation, inflammation and immune clearance [38]. Whilst they are considered a component of the innate immune system, they also act as an important bridge with the antibody dependent and cellular mediated immune responses. There are three characterised pathways of the complement system, including the classical pathway, the alternative pathway, and the mannose binding lectin pathway. The activation requirements of the respective pathways are different, but they all result in the production of anaphylatoxins ‘C3 and C5’ effector molecules and the membrane attack complex for cellular destruction. The activation of the classical pathway involves the binding of complement molecule ‘C1’ to functional regional region of an antibody currently bound to an antigen (i.e. antibody-antigen complex). On the other hand, the activation of the alternative pathway involves the direct binding of the complement molecule ‘C3’ to the microbial surface. Lastly, the activation of lectin pathway involves the activation of the complement molecule ‘C3’ through the binding of lectin to mannose on the microbial surface [39-41].
During cardiac surgery, complement is activated via blood surface interactions with extracorporeal circulation, ischemic reperfusion injury, and the reversal of heparinization [38, 42]. The adsorption of complement proteins (C3) to the extracorporeal circulation circuitry, results in the activation of the alternative complement pathway. Conversely, the neutralisation of heparin with protamine following the completion of cardiac surgery results in the formation of heparin-protamine complexes. These complexes have been shown to activate the complement cascade via the classic pathway [4]. In contrast, there is no clearly established mechanism to explain the predominant complement pathway activated in ischemia-reperfusion injury [43]. However, it is known that the complement protein C5a may facilitate ROS generation through the activation of xanthine dehydrogenase and the activation of membrane attack complexes [38, 44].
Most of the current literature suggest that increased complement levels following cardiac surgery are correlated with poorer clinical outcomes. Holmes et al 2002 observed that increased levels of C3a was associated with increased postoperative bleeding and delayed extubation times [44]. Weisman et al. 1990 demonstrated that the use of a complement activator inhibitor may significantly reduce myocardial necrosis after ischemic-reperfusion injury [45]. Recently, a C5 binding monoclonal antibody, Pexelizumab, has been developed. A phase II trial suggested that it is effective at reducing both 30 and 180 day mortality rates compared to placebo [46].
The coagulation cascade
The coagulation cascade consists of an intrinsic and extrinsic pathway, which converge onto a common pathway leading to thrombin formation and the production of an insoluble fibrin clot. The intrinsic pathway is activated by exposed endothelial collagen following tissue damage. Conversely, the extrinsic pathway is activated by tissue trauma, which facilitates the release of tissue factor [47]. The coagulation cascade is linked with inflammation, as activated platelets release inflammatory cytokines and facilitate leukocyte activation [48].
There are conflicting theories describing the activation of the coagulation cascade during CPB. It was thought that the intrinsic pathway is predominantly activated following the contact of factor XII against the CPB circuitry. However, Burman et al. 1994 reported that patients with factor XII deficiency were still able to generate thrombin [49]. In contrast, Somer et al. 2002 observed that tissue factor is the main activator of the coagulation cascade during CPB [50].
Cardiac surgery promotes a prothrombotic state. The intra-operative use of heparin minimises blood coagulation within the CPB circuit but does not prevent it completely [51]. Hence, thrombin production still occurs due to vascular injury from CPB, resulting in the consumption of clotting factors and coagulopathy following surgery [52].
Cytokines
Cytokines are signalling molecules with pro-inflammatory or anti-inflammatory characteristics. They are predominantly produced by macrophages, monocytes and lymphocytes [53, 54]. Cytokines act on a range of immunological mediators such as leukocytes or the endothelium. They may act locally, or travel via the systemic circulation or lymphatic system to exert their effects. The surgically relevant pro-inflammatory cytokines include interleukin-6 (IL-6), interleukin-8 (IL-8) and tumour necrosis factor a (TNFα) [53]. In contrast, the important surgical anti-inflammatory cytokines include interleukin-10 (IL-10) and interleukin-1 receptor antagonist (IL-1ra) [55].
In cardiac surgery, the pro-inflammatory response often over-rides the anti-inflammatory response. The level of serum IL-6 is increased during CPB and remains elevated for 48 hours postoperatively [56]. IL-6 is the predominant activator of the acute phase response during inflammation [57]. The level of IL-6 may be used to predict outcomes in patients undergoing cardiac surgery. Bairer et al. 2021 demonstrated that increased levels of IL-6 is associated with increased mortality and prolonged ICU admission following cardiac surgery [58]. Similarly, Zhang et al. 2015 suggests that increased levels of plasma IL-6 may predict acute kidney injury (AKI) after cardiac surgery [59]. Hirai et al. 2003 observed that the duration of SIRS significantly correlated with the level of IL-6 and duration of the aortic cross clamp time [60]. Additionally, IL-8 acts as a potent neutrophil chemoattractant and may contribute to myocyte injury [60]. Mehmet et al. 1995 demonstrated IL-8 may facilitate myocyte injury following prolonged cardiac ischemia, while Frering et al. 1994 suggests that IL-8 may play a crucial role in the pathophysiology of ischemic-reperfusion injury [61, 62]. TNFαis involved in stimulating neutrophil functions such as the generation of ROS and trans-endothelial migration [63]. However, there are conflicting findings regarding the TNFαresponse following cardiac surgery [53]. For example, Ozawa et al. 2000 demonstrated a significant increase in TNFαfollowing cardiac surgery, whereas Finn et al. 1993 did not [64, 65].
Neutrophils
Neutrophils, also known as polymorphonuclear cells, are the hallmark of acute inflammation [66]. As a crucial component of the innate immune system, they are responsible for phagocytosis of invading organisms, formation of extracellular traps, degranulation and production of reactive oxygen species [67]. Upon degranulation, neutrophils release elastase and myeloperoxidase, which function to destroy microbial features such as virulence factors and cell walls [68]. Furthermore, during inflammation, neutrophils are capable of quickly migrating to the insulted area by following chemotactic signals and cytokines. Cytokines upregulate the expression of adhesion molecules such as selectin and integrins on the endothelium, allowing neutrophils to migrate to the desired area [69].
Cardiac surgery with CPB is associated with an increased systemic concentration of elastase and myeloperoxidase, indicating there is increased neutrophil activity [70]. Neutrophils have been described as the potential effectors of ischemic reperfusion injury following CPB and appear to be drive pulmonary dysfunction [70, 71]. Additionally, neutrophil priming may occur after CPB because of mechanical sheer stress, resulting in an enhanced sensitivity to future stimuli. This decreases the threshold for systemic inflammation when challenged by future insults [72].
End Organ Dysfunction due to Cardiac Surgery Related Inflammation
Endothelial dysfunction
The endothelium is important for regulating vascular tone, the coagulation cascade, and trans-endothelial migration of neutrophil during inflammation [73]. Vascular tone is tightly controlled via factors including nitric oxide, endothelin, endothelium derived hyperpolarising factor and prostacyclin [74, 75]. The endothelium is an important mediator in the coagulation cascade, given tissue injury may lead to the activation of the intrinsic and extrinsic pathways [76]. As previously stated, trans-endothelial migration of neutrophils is achieved through the upregulation of adhesion molecules on the endothelial surface in response to pro-inflammatory cytokines such as selectin and integrins [69].
Following CPB, there is significant endothelial dysfunction in the pulmonary, cerebral and myocardial vessels. A pro-inflammatory state induced by CPB results in the generation of pro-inflammatory cytokines such as IL-1B and TNFα, which inhibit the production and bioavailability of endothelial vasodilators such as nitric oxide [4]. This commonly manifests as pulmonary hypertension due to an increase in pulmonary vascular resistance [77]. This is confirmed by an improvement in the pulmonary vascular resistance following the supplementation of nitric oxide [78]. Furthermore, microvascular dysfunction is associated with ischemic reperfusion injury. The balance between endothelium dependent vasodilation and vasoconstriction is shifted towards excessive vasoconstriction through the production of endothelin-1 and ROS, resulting in the exacerbation of tissue ischemia. Endothelial dysfunction may also induce platelet aggregation, promoting a prothrombotic state [33, 79]. From a different perspective, the importance of the endothelium is highlighted by the superior patency of the internal mammary artery compared to other conduits such as the saphenous vein used for CABG. The internal mammary artery releases higher levels of endothelium relaxing factors such as nitric oxide, contributing to a longer patency and duration of use [73].
Cardiovascular dysfunction
Major cardiovascular complications occur in at least 10% of patients undergoing coronary artery bypass grafting (CABG), including myocardial infarction, heart failure and cardiac death. These cardiovascular complications cost the United States an additional $2 billion USD per annum [80].
Cardiovascular dysfunction following CPB is largely due to ischemic-reperfusion injury [4]. This may clinically manifest as perioperative myocardial infarction, low cardiac output, arrhythmias and myocardial stunning [34]. Weman et al. 2000 demonstrated that ischemic-reperfusion injury caused a quarter of patient deaths following CABG via histological confirmation of contraction band necrosis on myocytes [81]. Similarly, Moore et al. 1981 demonstrated the presence of contraction band necrosis in myocytes in 45% of patients who underwent CABG (82). In a neonatal porcine model, piglets undergoing CPB had a larger decrease in cardiac output and poorer oxygenation compared to the piglets undergoing sham surgery. These piglets also demonstrated an increased accumulation of neutrophils within the myocardium [83].
Pulmonary dysfunction
Acute lung injury and acute respiratory distress syndrome are common manifestations following cardiac surgery, characterised by hypoxemia, bilateral pulmonary infiltrates, and normal pulmonary capillary wedge pressure [4, 84]. Pulmonary dysfunction post cardiac surgery may also manifest as increased vascular resistance, reduced pulmonary compliance, increased arteriovenous oxygen gradients and leakage of fluid into the interstitium [55]. The mortality rate of acute respiratory distress syndrome is 40% in the general population, but doubles to 80% in the cardiac surgery population [85].
The currently accepted pathophysiological mechanism of pulmonary dysfunction post cardiac surgery is ischemic-reperfusion injury and leucosequestration of neutrophils into the pulmonary parenchyma [70]. This is supported by the increased concentration of elastase released by neutrophils within the human pulmonary parenchyma, where it may exert its proteolytic activity [86, 87]. Furthermore, polymorphisms in human IL-6 and IL-18 genes have recently been demonstrated to increase the susceptibility of developing pulmonary dysfunction following cardiac surgery, reinforcing the deleterious effect of a systemic pro-inflammatory state [88, 89]. Animal models support the involvement of neutrophils in pulmonary dysfunction following CPB. Lung biopsies obtained from a porcine model subjected to CPB revealed abundant neutrophils, intra-alveolar haemorrhage and oedema [90]. Additionally, the use of an endothelial adhesion monoclonal antibody reduced the adhesion of neutrophils to the lung parenchyma in a rabbit model during ischemic reperfusion injury [91].
Renal dysfunction
Renal dysfunction such as AKI occurs in up to 18% of patients following cardiac surgery and is associated with significant morbidity and mortality [92]. A longer duration of CPB is associated with an increased incidence of renal dysfunction [93]. The extent of renal damage may potentially be predicted by an increase in renal tubular enzymes and an increased concentration of elastase [94]. Numerous mechanisms may contribute to renal dysfunction following cardiac surgery. The pro-inflammatory state induced by CPB results in the release of cytokines such as TNFα, which promotes the deposition of inflammatory cells and fibrin within the renal tubular cells and glomerulus. This results in local vasoconstriction and renal cell death, reducing the glomerular filtration rate [95]. Additionally, CPB is associated with the depletion of antioxidants in renal tubular cells, allowing reactive oxygen species to accumulate and damage renal epithelial cells [96]. Ischemic renal injury may also increase the sensitivity of the renal vasculature to vasoconstrictive stimuli, further reducing renal perfusion [97].
Cerebral dysfunction
The incidence of focal neurological defects such as transient ischemic attacks and perioperative stroke range between 1-5% following cardiac surgery. In contrast, up to 80% of patients experience cognitive decline post cardiac surgery [98]. Unfortunately, the underlying mechanism leading to cerebral dysfunction following cardiac surgery is poorly understood. Micro and macroembolization is one proposed mechanism of cerebral injury. It has been suggested that emboli composed of lipids originating from pericardial aspirate are returned to the CPB circuitry, where they may be distributed to the cerebral vasculature [99]. Additionally, emboli may develop from post-operative arrhythmias such as atrial fibrillation [100]. In contrast, cerebral dysfunction may alternatively occur via ischemic-reperfusion injury, with a 4x increase in the likelihood of cerebral dysfunction when patients experience a drop in systolic blood pressure below 50 mmHg for greater than 10 minutes during surgery [101]. The hypoperfusion of the brain may result in reduced clearance of emboli, leading to cerebral dysfunction [102, 103]. Furthermore, ischemic reperfusion injury may damage the blood-brain barrier, resulting in the passage of inflammatory mediators and oedema [104, 105].
Alkaline Phosphatase
Basics
Alkaline phosphatases (AP) are a family of metalloproteinases conserved across different species including bacteria and humans [106]. There are currently four characterised isoforms in humans, including tissue nonspecific AP (TNAP) and three tissue specific isoforms. The three tissue specific isoforms include intestinal, placental, and germ-cell isoforms. Tissue non-specific AP are the predominant isoform, and are typically found in the bone, liver and kidneys [107, 108].
Structure
AP is physiologically active as a homodimer, with each monomer consisting of 524 amino acid residues. AP’s active site consists of three metallic ions, including a Magnesium (Mg2+) ion, and two Zinc (Zn2+) ions, as well as a Serine (S) residue which acts as the binding site for phosphate molecules [107, 109]. AP is anchored to the cell membrane via glycosylphosphatidylinositol (GPI) [110]. A three-dimensional crystal structure of AP is depicted in Figure 2.
Function
Many physiological functions of AP are yet to be elucidated. However, the most well-known function of AP arises from the actions from tissue nonspecific AP found within the bone. Tissue non-specific AP found within the bone is responsible for bone mineralisation, increasing the availability of phosphate to allow for the deposition of extracellular matrix (hydroxyapatite) [107]. The intestinal form of AP is likely to facilitate intestinal absorption [106]. The intestinal, placental, and liver isoforms of AP may promote barrier defence by sitting outside cell membranes via the GPI anchor. When the inflammatory immune response is stimulated, AP is upregulated and cleaved from its GPI anchor on the cell membrane and subsequently released into systemic circulation [108, 110, 112]. AP can dephosphorylate one to two phosphates from the Lipid A group of LPS. LPS induced inflammation is attenuated by AP in several in vivo models. For example, Buemer et al. 2003 demonstrated that in mice affected by LPS sepsis, there was an 80% survival rate associated with the administration of calf intestinal AP [113]. Similarly, Bentala et al. 2002 observed that the administration of placental human AP resulted in a 100% survival rate in mice challenged with LPS [114].
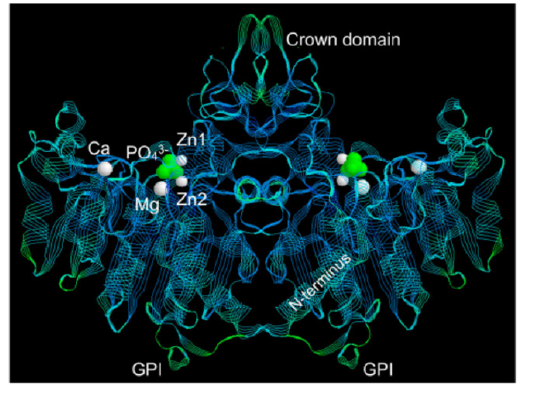
Figure2:A three-dimensional crystal structure of Alkaline Phosphatase, a homodimer. The active site is depicted in fluorescent-green and consists of three metallic ions including two Zinc (Zn2+) ions and one Magnesium (Mg2+) ion. The glycosylphosphatidylinositol (GPI) molecule is responsible for anchoring alkaline phosphatase to the cell membrane. Image by Millian et al 2016. (111)
Polestra et al. 1997 postulated that AP may also dephosphorylate extracellular nucleotides such as adenosine triphosphate (ATP), adenosine diphosphate (ADP) and adenosine monophosphate (AMP) into non-inflammatory nucleosines [108, 109, 115]. Intracellularly, the hydrolysis of ATP, ADP and AMP provide energy for physiological processes. However, when these molecules are found extracellularly, they are pro-inflammatory, induce vasoconstriction and promote platelet aggregation [116, 117]. This theory was confirmed by Davidson et al. 2016, who analysed the relationship between AP activity and 13C5-adenosine (radioactive form of adenosine) levels in infants undergoing CPB. They observed an association between the activity of AP and the production of 13C5-adenosine. When AP was challenged with a higher serum level of 13C5-AMP, a potential substrate, there was increased production of 13C5-adenosine (r=0.84; p<0.0001). In contrast, when AP was challenged with a lower level of 13C5-AMP, there was reduced production of 13C5-adenosine (r=0.50; p<0.05). The supplementation of AP has also been shown to significantly increase the production of 13C5-adenosine (p<0.0001) [116]. These findings suggest that AP can dephosphorylate both endotoxins and extracellular nucleotides, and the activity of AP is increased when there is an increased concentration of substrates.
Davidson et al. 2016 demonstrated that AP is likely to be the main enzyme to convert extracellular AMP into adenosine. Previously, CD73 has been reported as the main enzyme to convert AMP into adenosine (118). Therefore, to identify whether AMP or CD73 is the main enzyme, a TNAP inhibitor (MLS-0038949) and CD73 inhibitor was used on several post-operative samples. The TNAP inhibitor was significantly more effective at reducing the production of 13C5-adenosine when compared to no inhibitor (2.7 umol/L vs 11.9umol/L; p<0.0001). In contrast, the CD73 inhibitor was less effective at reducing the production of 13C5-adenosine when compared to no inhibitor (2.7 umol/L vs 8.3 umol/L; p<0.0001). Overall, these results suggest that AP is the main enzyme to dephosphorylate AMP, however, there were no direct comparisons drawn between the two inhibitors [116].
Loss of AP during cardiac surgery
Neutze et al. 1974 provided one of the initial studies to demonstrate that the level of AP decreases following cardiac surgery with CPB [119]. Lum et al. 1989 demonstrated a 48% reduction in AP activity in patients undergoing CPB compared to a 15% reduction in patients not undergoing CPB [120]. Kats et al. 2012 illustrated a reduction in the level of plasma AP within 2 hrs after the commencement of surgery, but this normalised by 24 hours [121]. Davidson et al. 2012 conducted a sub-analysis of a prospective observational study which aimed to analyse the kinetics of calcitonin in infants undergoing cardiac surgery. Patients were divided into the following intervention groups: “no CPB”, “CPB”, “delayed sternal closure”. A significant reduction in the level of AP was seen post-operatively in all groups but this loss was highest in the CPB and delayed sternal closure groups. Interestingly, patients undergoing cardiac surgery without CPB also demonstrated a reduction in the level of AP [122]. This suggests that the reduction in AP during cardiac surgery cannot be solely attributed to the use of CPB. Davidson et al. 2017 reinforced their previous findings, demonstrating that the level of AP is decreased following CPB (median decrease of 89 U/L; p<0.0001). However, they characterised that this loss is predominantly via the bone and liver-2 isoforms. The majority of AP loss occurred within 6 hours post-operatively. AP continued to decrease until 24 hours post-operatively in up to a third of patients, where it began to rise until the final measured timepoint of 72 hours post-operatively (P<0.0001) [109].
The mechanism underlying the loss of AP during cardiac surgery is poorly understood. Loss of AP during CPB because of the filtration system is unlikely to be culprit. The molecular size of AP (>65,000 Daltons) is larger than the CPB filter, which generally filters particles greater than 40 microns [122, 123]. Liver injury has also been proposed as a mechanism to explain the loss of AP during cardiac surgery [124]. However, Davidson et al. 2012 demonstrated that the level of AP was inversely related to the level of ALT. This contradicts the typical pattern of hepatocellular injury, where ALT should be decreased on liver function tests [122, 125]. Additionally, the consumption of AP to neutralise detrimental extracellular nucleotides generated by a systemic inflammatory response during CPB is a feasible hypothesis [110]. On the contrary, a reduction in the level of AP is still observed during off-pump cardiac surgery, suggesting that there are other causes for the loss of AP [109].
Safety profile of AP supplementation
Animal models of inflammation and sepsis provide key information regarding pharmacokinetics and the initial safety profile for drugs. Beumer et al. 2003 demonstrated that bovine AP attenuates the LPS mediated inflammatory response in piglets and mice by up to 80%. The dose of bovine AP up to 5354 U/KG was well tolerated [113]. There have been limited studies assessing the safety of AP supplementation in humans. Peters et al. 2015 conducted a phase 1, randomized, double blinded placebo-controlled trial to analyse the tolerability of recombinant AP (recAP). Recombinant AP was administered as a single, one-hour intravenous infusion (Doses varying from 200, 500, 1000 or 2000 U/kg; n = 33), or as multiple infusions (500 or 1000 U/kg; n=18), over three consecutive days. The drug was well tolerated, even at the highest dose of 2000 U/kg and serum antibodies against AP were not detected. The half-life of recombinant AP was long, ranging between 49-58 hours in the single infusion group, whilst ranging between 63-66 hours in the multiple infusion group. The serum concentration of recombinant AP peaked at the end of the 1-hour supplementation, before quickly declining to 10% of this value 4 hours post supplementation [126]. Pickers et al 2008 conducted a double blind, placebo-controlled study to examine the pharmacokinetics and pharmacodynamics of exogenously administered AP in both healthy and septic patients. They found exogenous AP follows linear pharmacokinetics. A stable level of serum AP may be achieved by administering a loading dose of 67.5 U/kg followed by a continuous IV infusion of AP within the range of 132.5 - 177.5 U/KG. An elimination half-life of 8-12 hrs was noted [127].
Clinical trials of alkaline phosphatase in cardiac surgery
The clinical trials using alkaline phosphatase during cardiac surgery have focussed on three broad areas; to assess the physiological implications of AP supplementation, the potential of AP to modulate the inflammatory response during cardiac surgery, and assess the clinical implications resulting from the loss of AP during cardiac surgery. A flow chart of the included studies is depicted in figure 3.
Supplementation of AP to increase De Novo synthesis
Kats et al. 2012 conducted a prospective randomised study of 63 patients undergoing CABG and demonstrated that supplementation of bovine intestinal AP (biAP) would induce the release of endogenous AP. Compared to the placebo group, the biAP supplementation group demonstrated a significant rise in the level of plasma AP 4-6 hours post-operatively (Pre-operative levels: 70.50 ±15.63 IU/L; 4-6 hrs Post Operatively: 354.97 ± 95 IU/L). This rise in plasma AP was confirmed to be caused by the de novo synthesis of AP as this peak is significantly larger than the administered dose. It was also established that the form of AP predominantly produced by the body during AP supplementation is tissue nonspecific AP, via the use of a tissue non-specific AP inhibitor (L-homoarginine) [121].
AP to modulate the inflammatory response during cardiac surgery
Kats et al. 2009 conducted a phase II double blind randomized study involving 63 patients to evaluate the anti-inflammatory effects of biAP compared to placebo in patients undergoing CABG with CPB (aka APIRRED Study). BiAP was administered as an intravenous bolus (1000 IU) prior to the commencement of surgery, followed by the immediate commencement of a continuous intravenous infusion (5.6 U/kg / hr) for 36 hours. Both the placebo and AP group exhibited a low inflammatory response, possibly due to a short aortic cross clamp time and the application of normothermia during CPB. However, five patients in placebo group had a large increase in the plasma concentration of TNFα which occurred 4 hours following the induction of surgery (Mean peak level: 108.1 ± 205.9 pg/ml). This was followed by an increase in the plasma level of other pro-inflammatory cytokines IL-6 (Mean peak: 682.6 ± 965.0 pg/ml) and IL-8 (Mean peak: 641.9 ± 836.7 pg/ml). In contrast, this large increase in pro-inflammatory cytokines was not recorded in the AP group (p<0.05). The patients enrolled in this study were lower risk surgical patients, as defined by a low to intermediate EUROSCORE [128].
Presbitero et al. 2018 produced a quantitative, validated model to analyse the effect of AP supplementation following cardiac surgery. They used data from clinical trials including the APIRRED trial discussed above. Their results revealed that AP supplementation results in a protective effect for cardiac surgery patients, via the de novo synthesis of AP [108].
Davidson et al. 2019 investigated whether there was an association between the serum level of AP and the clearance of endotoxins in 62 infants aged ≤ 120 days undergoing CPB. The level of endotoxins was measured using an endotoxin activity assay (EAA) pre-operatively, during the rewarming phase of CPB and one day post-operatively. Serum AP levels were measured at the same time points as for endotoxins above. The authors found that during CPB, serum AP levels were decreased by a mean of 94.8 U/L (P<0.001). Participants with a lower level of AP had a significantly higher levels of endotoxins measured pre-operationally (P<0.05) and during rewarming (P<0.01). Additionally, in 22 blood samples, the investigators examined whether the ex vivo supplementation of 1,600 U/L of human liver AP into pre-operative serum samples would reduce the level of endotoxins. Ex vivo supplementation of AP into pre-operative (mean EAA reduction of 29%; P<0.001) and post-operative (mean EAA reduction of 51%; P<0.0001) blood samples of patients who underwent CPB significantly reduced the levels of serum endotoxins. This study is one of the largest studies demonstrating the anti-inflammatory effects of AP in infants undergoing cardiac surgery [129].
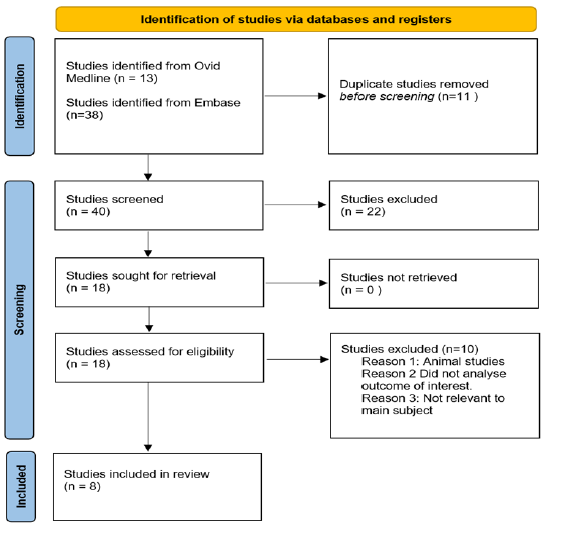
Figure3:Flow chart depicting the process of article inclusion for this review.
Alloksys Life Sciences is currently conducting a phase III study (APIRRED III) to assess whether the prophylactic administration of bovine intestinal AP can reduce the incidence and severity of AKI following cardiac surgery with CPB compared to placebo. AP will be administered as a bolus (1000 IU) at the start of the surgery, immediately followed by a large infusion (9000 IU) over 24 hours. Patients will then be followed up four weeks, assessing for the occurrence of AKI, length of hospital stay and mortality rates [130].
AP as a prognostic marker during cardiac surgery
Whilst the supplementation of AP during cardiac surgery is being explored for therapeutic purposes, changes in the level of AP may additionally be used as a prognostic factor. The loss of AP during cardiac surgery is correlated with poorer clinical outcomes. Through linear modelling, Davidson et al. 2012 illustrated that for each 10 U/L decrease in serum AP on post-operative day (POD) 1, there was a 6% increase in intubation times (p<0.001), a 5% increase in the length of hospital stays (p<0.05) and an increase in the vasoactive inotropic score at 24 hrs (VIS-24) by 0.7 (p<0.0001) (122). Davidson et al. 2017 demonstrated that low AP levels (defined as <80 U/L) at 72 hours post-operatively was associated with an increased odds of major cardiovascular events in infants, including cardiac arrest, unplanned post-operative mechanical circulatory support, or death within 30 days (OR 5.6; p<0.05) [109]. Schaefer et al. 2020 observed that a greater than 50% reduction in baseline level of AP following surgery was associated with a significantly higher hospital mortality, longer duration of intensive care and requirement for prolonged mechanical ventilation (defined as >24 hrs) (p<0.05). However, it should be noted that the same patients who experienced these large drops in AP required a longer duration of CPB and were more likely to require extracorporeal membrane oxygenation support, suggesting they may have more complicated disease (p<0.001) (110). Poschner et al. 2021 divided their patients based on the reduction of AP from baseline following surgery into ≥ 60% decrease, or <60% decrease. They found that patients with a ≥ 60% drop of AP following cardiac surgery were more likely to require renal replacement therapy (61.9% vs 40.9%; p=0.002), more likely to experience bleeding complications (60.0% vs 40.0%; p= 0.003) and have poorer outcomes at one year (p=0.031), compared to patients with a <60% drop in AP [131].
Conclusion
This review examines the inflammatory response to cardiac surgery, the current understanding of alkaline phosphatase and the potential of alkaline phosphatase a novel therapeutic agent to reduce inflammation post cardiac surgery. Alkaline phosphatase appears to be effective for reducing the inflammatory response during cardiac surgery in several clinic trials and is currently being examined in a phase III trial. If the efficacy of alkaline phosphatase is confirmed and its use is implemented into clinical practice, there would be a considerable reduction in the healthcare spending and patient morbidity due to post-operative complications resulting from inflammatory following cardiac surgery.
Conflicts of Interest
The authors declare no conflicts of interest.
Ethics
We believe to the best of our knowledge that the animal studies and clinical trials adhered to the ethical standards of the institutional and national guidelines.
Abbreviations
AP: Alkaline Phosphatase; AKI: Acute Kidney Injury; biAP: Bovine intestinal Alkaline Phosphatase; CAR: Counter Anti-inflammatory Response; CPB: Cardiopulmonary Bypass; DIC: Disseminated intravascular coagulation; EAA: Endotoxin Activity Assay; IL: Interleukin; LPS Lipopolysaccharide; POD: Postoperative day; ROS: Reactive oxygen species; SIRS: Systemic Inflammatory Response Syndrome; TNF: Tumour necrosis factor; TNAP: Tissue nonspecific alkaline phosphatase
References
1. Fuhrman DY, Kellum JA. Epidemiology and pathophysiology of cardiac surgery-associated acute kidney injury. Current Opinion Anesthesiol. 2017;30(1):60-5.
2. Natarajan A, Samadian S, Clark S. Coronary artery bypass surgery in elderly people. Postgrad Med J. 2007;83(977):154-8.
3. Rankin JA. Biological mediators of acute inflammation. AACN Advanced Crit Care. 2004;15(1):3-17.
4. Warltier DC, Laffey JG, Boylan JF, et al. The systemic inflammatory response to cardiac surgery: implications for the anesthesiologist. The J American Society of Anesthesiologists. 2002;97(1):215-52.
5. Boehne M, Sasse M, Karch A, et al. Systemic inflammatory response syndrome after pediatric congenital heart surgery: Incidence, risk factors, and clinical outcome. J Cardiac Surg. 2017;32(2):116-25.
6. Squiccimarro E, Labriola C, Malvindi P, et al. Prevalence and Clinical Impact of Systemic Inflammatory Reaction After Cardiac Surgery. J Cardiothorac Vasc Anesth. 2019;33.
7. Bone RC. Sir isaac newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24(7):1125-8.
8. Freeman BD, Natanson C. Anti-inflammatory therapies in sepsis and septic shock. Expert Opin Investig Drugs. 2000;9(7):1651-63.
9. Litmathe J, Boeken U, Bohlen G, et al. Systemic inflammatory response syndrome after extracorporeal circulation: a predictive algorithm for the patient at risk. Hellenic J Cardiol. 2011;52(6):493-500.
10. Bone RC. Gram-negative sepsis: background, clinical features, and intervention. Chest. 1991;100(3):802-8.
11. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-10.
12. Stevenson EK, Rubenstein AR, Radin GT, et al. Two decades of mortality trends among patients with severe sepsis: a comparative meta-analysis*. Crit Care Med. 2014;42(3):625-31.
13. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. The Lancet. 2020;395(10219):200-11.
14. Delano MJ, Ward PA. The immune system's role in sepsis progression, resolution, and long‐term outcome. Immunological reviews. 2016;274(1):330-53.
15. Schuler A, Wulf DA, Lu Y, et al. The impact of acute organ dysfunction on long-term survival among sepsis survivors. Critical care medicine. 2018;46(6):843.
16. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. The Lancet. 2018;392(10141):75-87.
17. Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36(1):296-327.
18. Comstedt P, Storgaard M, Lassen AT. The Systemic Inflammatory Response Syndrome (SIRS) in acutely hospitalised medical patients: a cohort study. Scand J Trauma Resusc Emerg Med. 2009;17:67-.
19. Kats S, Schönberger JP, Brands R, et al. Endotoxin release in cardiac surgery with cardiopulmonary bypass: pathophysiology and possible therapeutic strategies. An update. Eur j cardio-thorac surg. 2011;39(4):451-8.
20. Lin E, Gletsu-Miller N. Surgical Stress Induces an Amplified Inflammatory Response in Patients with Type 2 Diabetes. ISRN Obesity. 2013;2013:910586.
21. Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. 2007;38(12):1336-45.
22. Addonizio VP, Colman RW. Platelets and extracorporeal circulation. Biomaterials. 1982;3(1):9-15.
23. Edmunds Jr LH. Inflammatory response to cardiopulmonary bypass. Ann Thorac Surg. 1998;66(5):S12-S6.
24. Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71(1):635-700.
25. Andersen LW, Baek L, Degn H, et al. Presence of circulating endotoxins during cardiac operations. J Thorac Cardiovasc Surg. 1987;93(1):115-9.
26. Aydin NB, Gercekoglu H, Aksu B, et al. Endotoxemia in coronary artery bypass surgery: A comparison of the off-pump technique and conventional cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2003;125(4):843-8.
27. Rocke D, Gaffin S, Wells M, et al. Endotoxemia associated with cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1987;93(6):832-7.
28. Ohri SK, Bjarnason I, Pathi V, et al. Cardiopulmonary bypass impairs small intestinal transport and increases cut permeability. Ann Thorac Surg. 1993;55(5):1080-6.
29. Oudemans-van Straaten H, Hoek F, Van Deventer S, et al. Gastrointestinal permeability in cardiac surgery patients, the interpretation of dual-sugar absorption studies (multiple letters)[4]. Intensive care med. 1997;23(10):1101-2.
30. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229-317.
31. Gelman S. The pathophysiology of aortic cross-clamping and unclamping. The Journal of the American Society of Anesthesiologists. 1995;82(4):1026-57.
32. Larmann J, Theilmeier G. Inflammatory response to cardiac surgery: cardiopulmonary bypass versus non-cardiopulmonary bypass surgery. Best Pract Res Clin Anaesthesiol. 2004;18(3):425-38.
33. Verma S, Fedak PWM, Weisel RD, et al. Fundamentals of Reperfusion Injury for the Clinical Cardiologist. Circulation. 2002;105(20):2332-6.
34. Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010;106(3):360-8.
35. Bolli R, Jeroudi MO, Patel BS, et al. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci U S A. 1989;86(12):4695-9.
36. Lemasters JJ, Bond JM, Chacon E, et al. The pH paradox in ischemia-reperfusion injury to cardiac myocytes. Exs. 1996;76:99-114.
37. Bolli R. Mechanism of myocardial" stunning". Circulation. 1990;82(3):723-38.
38. Stahl GL, Shernan SK, Smith PK, et al. Complement activation and cardiac surgery: a novel target for improving outcomes. Anesth Analg. 2012;115(4):759-71.
39. Müller-Eberhard HJ. Molecular organization and function of the complement system. Annual review of biochemistry. 1988;57(1):321-47.
40. Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343(1):227-35.
41. Taylor P, Botto M, Walport M. The complement system. Current Biol. 1998;8(8):R259-R61.
42. Tarnok A, Hambsch J, Emmrich F, et al. Complement activation, cytokines, and adhesion molecules in children undergoing cardiac surgery with or without cardiopulmonary bypass. Pediatric cardiol. 1999;20(2):113-25.
43. Gorsuch WB, Chrysanthou E, Schwaeble WJ, et al. The complement system in ischemia-reperfusion injuries. Immunobiol. 2012;217(11):1026-33.
44. Holmes Iv J, Connolly N, Paull D, et al. Magnitude of the inflammatory response to cardiopulmonary bypass and its relation to adverse clinical outcomes. Inflammation Res. 2002;51(12):579-86.
45. Weisman H, Bartow T, Leppo M, et al. Recombinant soluble CR1 suppressed complement activation, inflammation, and necrosis associated with reperfusion of ischemic myocardium. Proc Assoc Am Physicians.1990;103:64-72.
46. Sellke FW, Boodhwani M. Inhibition of complement activation in cardiac surgery. J Thorac Cardiovasc Surg. 2006;131(2):266-7.
47. Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance, and regulation. Biochemistry. 1991;30(43):10363-70.
48. Schenone M, Furie BC, Furie B. The blood coagulation cascade. Current opinion in hematology. 2004;11(4):272-7.
49. Burman JF, Chung HI, Lincoln J, et al. Role of factor XII in thrombin generation and fibrinolysis during cardiopulmonary bypass. The Lancet. 1994;344(8931):1192-3.
50. De Somer F, Van Belleghem Y, Caes F, et al. Tissue factor as the main activator of the coagulation system during cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2002;123(5):951-8.
51. O'Carroll-Kuehn BU, Meeran H. Management of coagulation during cardiopulmonary bypass. Continuing education in anaesthesia, critical care & pain. 2007;7(6):195-8.
52. Höfer J, Fries D, Solomon C, et al. A snapshot of coagulopathy after cardiopulmonary bypass. Clin Appl Thromb Hemost. 2016;22(6):505-11.
53. Tønnesen E, Christensen VB, Toft P. The role of cytokines in cardiac surgery. Int J Cardiol 1996;53:S1-S10.
54. Lin E, Calvano SE, Lowry SF. Inflammatory cytokines and cell response in surgery. Surgery. 2000;127(2):117-26.
55. Brix‐Christensen V. The systemic inflammatory response after cardiac surgery with cardiopulmonary bypass in children. Acta Anaesthesiol Scand. 2001;45(6):671-9.
56. Butler J, Chong GL, Baigrie RJ, et al. Cytokine responses to cardiopulmonary bypass with membrane and bubble oxygenation. Ann Thorac Surg. 1992;53(5):833-8.
57. Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. 1990;265(3):621-36.
58. Bauer A, Korten I, Juchem G, et al. EuroScore and IL-6 predict the course in ICU after cardiac surgery. Eur J Med Res. 2021;26(1):29.
59. Zhang WR, Garg AX, Coca SG, et al. Plasma IL-6 and IL-10 Concentrations Predict AKI and Long-Term Mortality in Adults after Cardiac Surgery. J Am Soc Nephrol. 2015;26(12):3123-32.
60. Hirai S. Systemic inflammatory response syndrome after cardiac surgery under cardiopulmonary bypass. Ann Thorac Cardiovasc Surg. 2003;9(6):365-70.
61. Frering B, Philip I, Dehoux M, et al. Circulating cytokines in patients undergoing normothermic cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1994;108(4):636-41.
62. Oz MC, Liao H, Naka Y, et al. Ischemia-Induced Interleukin-8 Release After Human Heart Transplantation. Circulation. 1995;92(9):428-32.
63. Ferrante A. Activation of neutrophils by interleukins-1 and-2 and tumor necrosis factors. Immunol series. 1992;57:417-36.
64. Ozawa T, Yoshihara K, Koyama N, et al. Clinical efficacy of heparin-bonded bypass circuits related to cytokine responses in children. Ann Thorac Surg. 2000;69(2):584-90.
65. Finn A, Naik S, Klein N, et al. Interleukin-8 release and neutrophil degranulation after pediatric cardiopulmonary bypass. J. Thorac Cardiovasc Surg. 1993;105(2):234-41.
66. Soehnlein O, Steffens S, Hidalgo A, et al. Neutrophils as protagonists and targets in chronic inflammation. Nature Reviews Immunol. 2017;17(4):248-61.
67. Rosales C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front Physiol. 2018;9:113-.
68. Papayannopoulos V, Metzler KD, Hakkim A, et al. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677-91.
69. Wagner JG, Roth RA. Neutrophil migration during endotoxemia. Journal of leukocyte biology. 1999;66(1):10-24.
70. Tönz M, Mihaljevic T, von Segesser LK, et al. Acute lung injury during cardiopulmonary bypass. Are the neutrophils responsible? Chest. 1995;108(6):1551-6.
71. Butler J, Pathi VL, Paton RD, et al. Acute-phase responses to cardiopulmonary bypass in children weighing less than 10 kilograms. Ann Thorac Surg. 1996;62(2):538-42.
72. Kawahito K, Kobayashi E, Ohmori M, et al. Enhanced responsiveness of circulatory neutrophils after cardiopulmonary bypass: increased aggregability and superoxide producing capacity. Artif Organs. 2000;24(1):37-42.
73. He G-W. Vascular Endothelial Function Related to Cardiac Surgery. Asian Cardiovasc Thorac Ann. 2004;12(1):1-2.
74. Parkington HC, Coleman HA, Tare M. Prostacyclin and endothelium-dependent hyperpolarization. Pharmacol Res. 2004;49(6):509-14.
75. Tare M, Parkington HC, Coleman HA, et al. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346(6279):69-71.
76. The endothelium: vascular control of haemostasis. Eur J Obstet Gynecol. 2001;95(2):198-201.
77. Morita K, Ihnken K, Buckberg GD, et al. Pulmonary vasoconstriction due to impaired nitric oxide production after cardiopulmonary bypass. Ann Thorac Surg. 1996;61(6):1775-80.
78. Schulze-Neick I, Penny DJ, Rigby ML, et al., editors. L-arginine and substance P reverse the pulmonary endothelial dysfunction caused by congenital heart surgery. Critical Care. 1999: Springer.
79. Lefer AM, Tsao PS, Lefer DJ, et al. Role of endothelial dysfunction in the pathogenesis of reperfusion injury after myocardial ischemia. The FASEB J. 1991;5(7):2029-34.
80. Mangano DT. Cardiovascular Morbidity and CABG Surgery—A Perspective: Epidemiology, Costs, and Potential Therapeutic Solutions. J Card Surg. 1995;10(s4):366-8.
81. Weman SM, Karhunen PJ, Penttilä A. Reperfusion injury associated with one-fourth of deaths after coronary artery bypass grafting. Ann Thorac Surg. 2000;70(3):807-12.
82. Moore GW, Hutchins GM. Coronary artery bypass grafts in 109 autopsied patients. Statistical analysis of graft and anastomosis patency and regional myocardial injury. JAMA. 1981;246(16):1785-9.
83. Brix-Christensen V, Tønnesen E, Hjortdal VE, et al. Neutrophils and platelets accumulate in the heart, lungs, and kidneys after cardiopulmonary bypass in neonatal pigs. Crit Care Med. 2002;30(3).
84. Dushianthan A, Grocott MPW, Postle AD, et al. Acute respiratory distress syndrome and acute lung injury. Postgrad Med J. 2011;87(1031):612-22.
85. Rong LQ, Di Franco A, Gaudino M. Acute respiratory distress syndrome after cardiac surgery. J Thorac Dis. 2016;8(10):E1177-E86.
86. Ascione R, Lloyd CT, Underwood MJ, et al. Inflammatory response after coronary revascularization with or without cardiopulmonary bypass. Ann Thorac Surg. 2000;69(4):1198-204.
87. Ng CSH, Wan S, Yim APC, et al. Pulmonary dysfunction after cardiac surgery*. Chest. 2002 2002/04//:1269+.
88. Chen S, Xu L, Tang J. Association of interleukin 18 gene polymorphism with susceptibility to the development of acute lung injury after cardiopulmonary bypass surgery. Tissue Antigens. 2010;76(3):245-9.
89. Wang JF, Bian JJ, Wan XJ, et al. Association between inflammatory genetic polymorphism and acute lung injury after cardiac surgery with cardiopulmonary bypass. Med Sci Monit. 2010;16(5):Cr260-5.
90. Zehr KJ, Poston RS, Lee PC, et al. Platelet activating factor inhibition reduces lung injury after cardiopulmonary bypass. Ann Thorac Surg. 1995;59(2):328-35.
91. Adkins WK, Taylor AE. Role of xanthine oxidase and neutrophils in ischemia-reperfusion injury in rabbit lung. J Appl Physiol. 1990;69(6):2012-8.
92. Thiele RH, Isbell JM, Rosner MH. AKI Associated with Cardiac Surgery. Clin J Am Soc Nephrol. 2015;10(3):500-14.
93. Fischer UM, Weissenberger WK, Warters RD, Geissler HJ, Allen SJ, Mehlhorn U. Impact of cardiopulmonary bypass management on postcardiac surgery renal function. Perfusion. 2002;17(6):401-6.
94. Hashimoto K, Miyamoto H, Suzuki K, et al. Evidence of organ damage after cardiopulmonary bypass: the role of elastase and vasoactive mediators. J Thorac Cardiovasc Surg. 1992;104(3):666-73.
95. Meldrum DR, Donnahoo KK. Role of TNF in mediating renal insufficiency following cardiac surgery: evidence of a postbypass cardiorenal syndrome. J Surg Res. 1999;85(2):185-99.
96. Dennis JM, Witting PK. Protective Role for Antioxidants in Acute Kidney Disease. Nutrients. 2017;9(7):718.
97. KE, Chu MW, Novick RJ, et al. Perioperative N-acetylcysteine to prevent renal dysfunction in high-risk patients undergoing CABG surgery: a randomized controlled trial. Jama. 2005;294(3):342-50.
98. Arrowsmith JE, Grocott HP, Reves JG, et al Central nervous system complications of cardiac surgery. BJA: British Journal of Anaesthesia. 2000;84(3):378-93.
99. Brooker RF, Brown WR, Moody DM, et al. Cardiotomy suction: a major source of brain lipid emboli during cardiopulmonary bypass. Ann Thorac Surg. 1998;65(6):1651-5.
100. Mathew JP, Fontes ML, Tudor IC, et al. A multicenter risk index for atrial fibrillation after cardiac surgery. JAMA. 2004;291(14):1720-9.
101. Tufo HM, Ostfeld AM, Shekelle R. Central nervous system dysfunction following open-heart surgery. Jama. 1970;212(8):1333-40.
102. Caplan LR, Hennerici M. Impaired clearance of emboli (washout) is an important link between hypoperfusion, embolism, and ischemic stroke. Arch Neurol. 1998;55(11):1475-82.
103. Stump DA, Rogers AT, Hammon JW, et al. Cerebral emboli and cognitive outcome after cardiac surgery. J Cardiothorac Vasc Anesth. 1996;10(1):113-9.
104. Lin L, Wang X, Yu Z. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem Pharmacol (Los Angel). 2016;5(4):213.
105. Carbone F, Bonaventura A, Montecucco F. Neutrophil-Related Oxidants Drive Heart and Brain Remodeling After Ischemia/Reperfusion Injury. Front Physiol. 2020;10:1587-.
106. Millán JL. Alkaline Phosphatases : Structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006;2(2):335-41.
107. Ndrepepa G. Alkaline phosphatase and cardiovascular disease. J Lab Precis Med. 2017;2(10).
108. Presbitero A, Mancini E, Brands R, et al. Supplemented alkaline phosphatase supports the immune response in patients undergoing cardiac surgery: clinical and computational evidence. Front Immunol. 2018;9:2342.
109. Davidson JA, Urban TT, Baird C, et al. Alkaline phosphatase in infant cardiopulmonary bypass: kinetics and relationship to organ injury and major cardiovascular events. J Pediatr. 2017;190:49-55. e2.
110. Schaefer A-K, Hutschala D, Andreas M, et al. Decrease in serum alkaline phosphatase and prognostic relevance in adult cardiopulmonary bypass. Interact Cardiovasc Thorac Surg. 2020;31(3):383-90.
111. Millán JL, Whyte MP. Alkaline phosphatase and hypophosphatasia. Calcified tissue international. 2016;98(4):398-416.
112. Kaliannan K, Hamarneh SR, Economopoulos KP, et al. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. PNAS. 2013;110(17):7003-8.
113. Beumer C, Wulferink M, Raaben W, et al. Calf intestinal alkaline phosphatase, a novel therapeutic drug for lipopolysaccharide (LPS)-mediated diseases, attenuates LPS toxicity in mice and piglets. J Pharmacol Exp Ther. 2003;307(2):737-44.
114. Bentala H, Verweij WR, Huizinga-Van der Vlag A, et al. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock. 2002;18(6):561-6.
115. Poelstra K, Bakker WW, Klok PA, et al. A physiologic function for alkaline phosphatase: endotoxin detoxification. Lab Invest. 1997;76(3):319-27.
116. Davidson JA, Urban T, Tong S, et al. Alkaline phosphatase, soluble extracellular adenine nucleotides, and adenosine production after infant cardiopulmonary bypass. PLoS One. 2016;11(7):e0158981.
117. Peters E, Masereeuw R, Pickkers P. The Potential of Alkaline Phosphatase as a Treatment for Sepsis-Associated Acute Kidney Injury. Nephron Clin Pract. 2014;127(1-4):144-8.
118. Pettengill M, Robson S, Tresenriter M, et al. Soluble ecto-5′-nucleotidase (5′-NT), alkaline phosphatase, and adenosine deaminase (ADA1) activities in neonatal blood favor elevated extracellular adenosine. J Biol Chem. 2013;288(38):27315-26.
119. Neutze J, Drakeley M, Barratt-Boyes B, et al. Serum enzymes after cardiac surgery using cardiopulmonary bypass. Am Heart J. 1974;88(4):425-42.
120. Lum G, Marquardt C, Khuri SF. Hypomagnesemia and low alkaline phosphatase activity in patients' serum after cardiac surgery. Clin Chem. 1989;35(4):664-7.
121. Kats S, Brands R, Hamad MAS, et al. Prophylactic treatment with alkaline phosphatase in cardiac surgery induces endogenous alkaline phosphatase release. Int J Artif Organs. 2012;35(2):144-51.
122. Davidson J, Tong S, Hauck A, et al. Alkaline phosphatase activity after cardiothoracic surgery in infants and correlation with post-operative support and inflammation: a prospective cohort study. Critical Care. 2012;16(4):1-10.
123. Groom RC, Quinn RD, Lennon P, et al. Detection and elimination of microemboli related to cardiopulmonary bypass. Circ Cardiovasc Qual Outcomes. 2009;2(3):191-8.
124. Sabzi F, Faraji R. Liver Function Tests Following Open Cardiac Surgery. J Cardiovasc Thorac Res. 2015;7(2):49-54.
125. Giannini EG, Testa R, Savarino V. Liver enzyme alteration: a guide for clinicians. CMAJ. 2005;172(3):367-79.
126. Peters E, Arend J, Tiessen R, et al. Pharmacokinetics, safety and tolerability of human recombinant alkaline phosphatase in healthy volunteers. Critical Care. 2015;19(1):1-201.
127. Pickkers P, Snellen F, Rogiers P, et al. Clinical pharmacology of exogenously administered alkaline phosphatase. Eur J Clin Pharmacol. 2009;65(4):393-402.
128. Kats S, Brands R, Seinen W, et al. Anti-inflammatory effects of alkaline phosphatase in coronary artery bypass surgery with cardiopulmonary bypass. Recent Pat Inflamm Allergy Drug Discov. 2009;3(3):214-20.
129. Davidson JA, Urban TT, Tong S, et al. Alkaline phosphatase activity and endotoxemia after infant cardiothoracic surgery. Shock (Augusta, Ga). 2019;51(3):328.
130. Preventing Oxidative stress induced Ischemic Injury- and Systemic Inflammation complications during and after invasive Cardiac surgery with Alkaline Phosphatase (APPIRED III) [Internet]. EU Clinical Trials Register. 2021 [cited 26/06/2021]. Available from: https://www.clinicaltrialsregister.eu/ctr-search/trial/2016-002663-33/AT#A.
131. Poschner T, Schaefer A-K, Hutschala D, et al. Impact of Venoarterial Extracorporeal Membrane Oxygenation on Alkaline Phosphatase Metabolism after Cardiac Surgery. Biomolecules. 2021;11(5):748.
Received: July 01, 2021.
Accepted: July 23, 2021.
Published: July 31, 2021.
To cite this article : Vu T, Fricke TA, Smith JA. A Review of Alkaline Phosphatase in Preventing Systemic Inflammation after Cardiac Surgery. European Journal of Respiratory Medicine. 2021; 3:2.
© 2021 Vu T, et al.