
Research Article/ Open Access
DOI:10.31488/EJRM.126
Crizotinib Restored Pembrolizumab Response in Advanced Non-Small Cell Lung Cancer
Laila C. Roisman#1, Nir Peled*1, Kiran Kundu2,3, Susmita Ghosh3, Bilal Krayim1, Jonah M. Cooper4, Walid Shalata5, Yehonatan Aharon Cohen5, Moshe Elkabets2, Angel Porgador2,3, Dina Levitas1, Waleed Kian#1, Ariel Sobarzo#2
1.The Oncology Institute, Shaare Zedek Medical Center, Jerusalem, Israel
2.The Shraga Segal Department of Microbiology, Immunology, and Genetics, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer Sheva, Israel
3.National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Beer Sheva, Israel
4.Medical School for International Health, Ben-Gurion University of the Negev, Beer Sheva, Israel
5.The Legacy Heritage Oncology Center and Dr. Larry Norton Institute, Soroka Medical Center and Ben-Gurion University, Beer-Sheva, Israel
*Corresponding author: Prof. Nir Peled, MD PhD FCCP, Head of Oncology Division, Shaare Zedek Medical Center, 12, Shmuel Beit St, Jerusalem, Israel 9103102, Tel: +972 (0)58 7040620
*Equal Contribution: Dr. Roisman and Prof. Peled contributed equally to this work and should be considered as a first author. Dr. Kian and Sobarzo contributed equally to this work and should be considered as a last author.
Abstract
Background: PD-1/PD-L1 immune checkpoint inhibitors, such as pembrolizumab and nivolumab, are used as a backbone treatment in various types of cancers, including lung cancer. However, PD‑1/PD-L1 inhibitors effect a small number of patients, and acquired resistance to those treatments is still unresolved. Furthermore, the identification of potential anti‑PD‑1/PD-L1 sensitizers for restoring treatment response and effectiveness is a challenge. Materials and Methods: In this report, we presented a patient with non-small cell lung cancer (NSCLC) who received repeated rounds of pembrolizumab and showed a prolonged effective response, although eventually, he developed resistance to this treatment course. Results: Notably, this patient developed acquired resistance to PD-1/PD-L1, which was associated with several driver gene alternations, including a de novo profound MET overexpression. In vitro studies using the patient tumor cells and CD8+ T cells revealed significant reactivation and resensitization to pembrolizumab upon combined treatment with crizotinib. Combined treatment of crizotinib and pembrolizumab in this patient resulted in a significant clinical and RECIST response. Conclusions:
Treatment strategy in patients with PD-1/PD-L1 acquired resistance should routinely include comprehensive genotyping of targetable driver oncogenes such as MET overexpression, along with the determination of PD-L1 status. These driver gene mutations may provide a vital target for immune checkpoint inhibitors therapy resensitization and optimize treatment stratification.
Keywords: MET, Crizotinib, Pembrolizumab, Lung cancer, NSCLC
Introduction
Immune checkpoint inhibitors (ICI) such as pembrolizumab, a programmed death/ligand 1 (PD-1/PD-L1) blocker, works by disrupting PD-1/PD-L1 direct interactions in the tumor microenvironment. In clinical practice, anti-PD-1/PD-L1 antibodies treatment has resulted in durable tumor remission, and they are routinely used to treat non-small cell lung cancer (NSCLC) patients [1–3]. Despite relative success, pembrolizumab therapy is hampered by primary and acquired resistance [4], and disease progression or relapse occurs even after a complete response has been achieved [5]. The reasons for that are still not fully understood [6]. Underlying resistance mechanisms are a large range of tumor factors, including genomic features, transcriptomic signatures, and immune landscape, to host factors. Acquired resistance is mainly associated with loss of the neoantigen and deficiency in its presentation, loss of T-cell effector function, and up-regulation of alternate immune checkpoint receptors [7,8]. There is an urgent need to develop approaches to improve the sensitivity of PD‑1/PD‑LI inhibitors [9], and focus efforts towards identifying a new combination therapy with PD-1/PD-L1 inhibitors to provide better therapeutic strategies [9,10].
Studies have shown that several tumor-related gene alterations in NSCLC are associated with controlling the anti-tumor immune response [11]. These alterations were suggested to facilitate tumor growth by enabling tumor cells to escape immune surveillance [11]. For example, a genetic alteration is the mesenchymal-epithelial transition receptor (MET) [12]. MET is a tyrosine kinase receptor primarily found on epithelial cells, regulating cell motility and proliferation [13,14]. Abnormal MET signaling, including MET receptor overexpression, receptor mutation, or amplification, are observed in NSCLC [15,16]. De novo MET gene amplifications are found in 1% to 5% of NSCLC cases adenocarcinoma, and MET overexpression occurs in 35% to 72% of NSCLC cases [17,18]. The activation of MET regulates distinct downstream signaling [19]. This signaling regulates cell survival and proliferation, migration and invasion angiogenesis, and epithelial to mesenchymal cell transformation. MET is also shown to have significant immunosuppressive roles [12,20,21].
Here, we report a case with advanced metastatic NSCLC, who presented with a resistance to a PD-1/PD-L1 therapy associated with tumor genetic alternations, notably an exceptionally high MET overexpression. In vitro studies using CD8+ T cells and tumor cells collected from this patient during disease progression and treatment resistance showed robust reactivation and significant resensitization to pembrolizumab upon the combination with crizotinib.
Case Presentation
In January 2015, a 62-year-old male with previous smoking history was admitted to our hospital. He was diagnosed with metastatic NSCLC. Molecular analysis (FoundationOne) from supraclavicular lymph node biopsy revealed ERBB2 amplification and gene alterations; VHL(Q195R), GRM3 (E517K), TP53 (R282W), FAT1 (Q4458), and SMAD4 (Q245). No mutation was found in EGFR, ALK, ROS1, RET, KRAS, and MET genes (Figure 1). Based on the molecular findings, the patient received two cycles of first-line chemotherapy of carboplatin (AUC=6, 2 cycles every 3 weeks) and paclitaxel (200mg/m2 every 3 weeks) and radiotherapy (chemoRT) treatment (total dose 63Gy), with adaptational stereotactic radiosurgery (SRS) for his brain metastases (total dose 20Gy). However, post-treatment laboratory findings were notable for elevated liver function test due to multiple liver metastases. In November 2015, the molecular analysis of biopsy collected in January 2015 showed both high PD-L1 expression (>90%) and a high Tumor Mutational Burden (TMB) of 38 (FoundationOne). Based on these new results, second-line pembrolizumab (3 cycles of 200mg every 3 weeks) was administrated for three months. PET-CT scan in March 2016 demonstrated a continuing worsening of the disease, with suspected metastatic lesion appearance in the abdomen and clinical signs including severe pain and weight loss (Figure 2A-B). The clinical changes reported by the patients were interpreted as complications of radiotherapy potentiated by the pembrolizumab treatment. Therefore, pembrolizumab was discontinued. After three months of supportive care, the patient restarted a pembrolizumab treatment course, which had a clinically significant response that lasted for ten months (Figure 2C). In April 2017, Magnetic Resonance Imaging (MRI) showed severe brain necrosis and edema (Figure 2F). Pembrolizumab treatment was discontinued due to immune-related adverse events (IRAEs). These included asthenia, fatigue, nausea, and diarrhea associated with ICI therapy [11]. A high dose of a steroid, methylprednisolone (2mg/kg every day for 5 days), combined with bevacizumab (150mg every 3 weeks) was administrated.
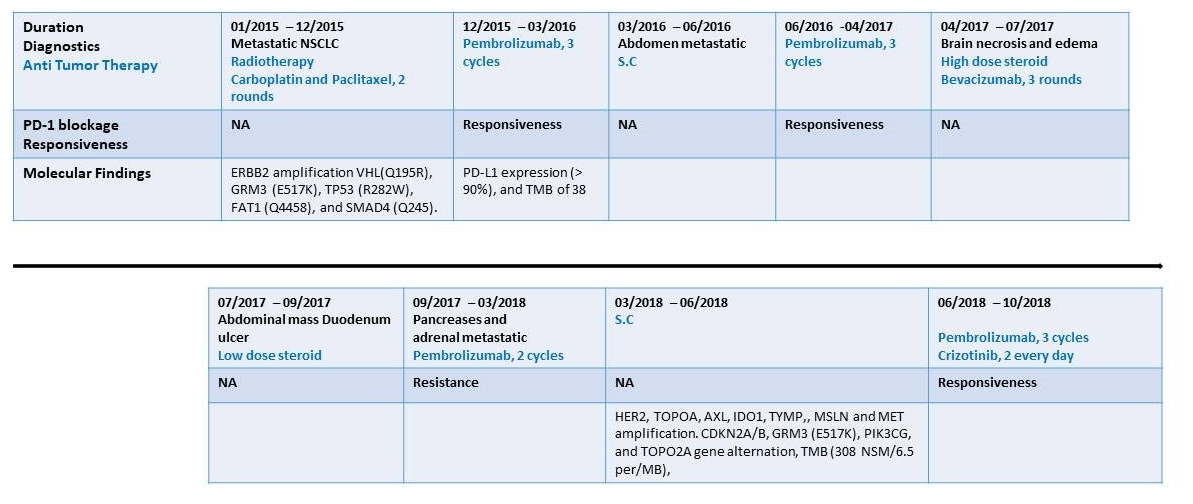
Figure 1:Time axis of diagnostics, anti-tumor treatment and intervention and PD-1 blockadge treatment response. Line graph illustrating disease progression, anti-tumor therapy and PD-1 blockage (pembrolizumab) treatment response from January 2015 to October 2018. NA – Not applicable; NSCLC- Non small cell lung cancer, NSM - Non-Synonymous Mutation
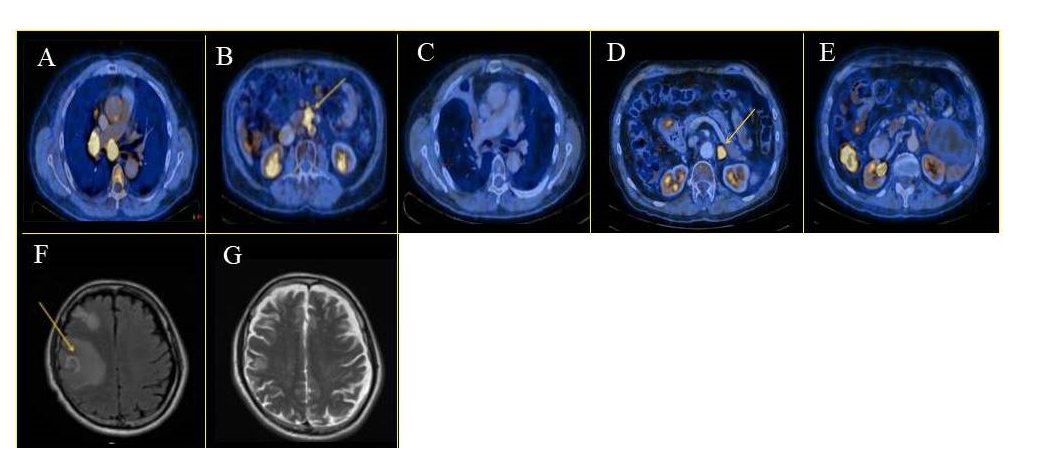
Figure 2:Anti tumor treatment response. PET-CT and MRI images of the patient treatment response at various stages of disease progression. (A) Abdominal metastases without pembrolizumab treatment, January 2015. (B) Abdomen FDG uptake, after three months of pembrolizumab treatment, significant clinical improvement, March 2016. (C) The patient continues pembrolizumab treatment and showed a positive clinical response, January 2017. (D) Single left adrenal mass after five months without pembrolizumab treatment. The patient was re-challenged with pembrolizumab and showed no response to treatment, September 2017. (E) Following new molecular and the detection of de novo MET overexpression, the patient received combined crizotinib and pembrolizumab treatment, resulting in significant clinical improvement. (F) Neurological symptoms, severe Rt. Hemisphere edema and necrosis. Pembrolizumab treatment was stopped. April 2017. (G) Neurological improvement with edema reduction after steroids and bevacizumab treatment. Yellow arrow indicates lesions and metastases.
Follow-up MRI, three months later, showed significant clinical improvement and reduction of cerebral edema (Figure 2G); however, the patient complained about severe abdominal pain. Endoscopic examination revealed a drug-induced ischemic ulcer in the upper duodenum. Bevacizumab treatment was stopped, and a lower dose of steroids was administrated. The patient showed clinical improvement for the following two months.
In September 2017, PET-CT revealed oligometastatic progression in the left pancreas and adrenal gland. The patient declined radiotherapy, and therefore, a re-challenge of pembrolizumab was initiated (3 cycles of 200 mg every 3 weeks). The patient tumor was not responsive to this regimen, and disease progression was observed (Figure 2D). Accordingly, metastasectomy was performed, requiring prolonged post-operative support because of local complications. He continued with supportive care, and seven months after surgery, PET-CT scans confirmed disease progression with numerous abdominal metastases. Updated molecular analysis (NathHealth) in March 2018 of a second biopsy specimen, taken from an adrenal gland lesion at the time of resistance to pembrolizumab, revealed an increase in mutation allele frequency of the previously altered genes (detected in the 2015 biopsy). Additional new alterations were found in several genes; CDKN2A/B, GRM3 (E517K), PIK3CG, and TOPO2A. Expression analysis (GPS Cancer test; NathHealth) showed high TMB (308 non-synonymous mutation/6.5 per/MB), and high expression levels of MET, a de novo MET gene copy number (x8) and protein overexpression (2239 mmol/µg).and elevated levels of HER2, TOPOA, AXL, IDO1, TYMP, and MSLN. In vitro studies using the patient tumor cells and CD8+ T cells revealed robust reactivation and resensitization to pembrolizumab upon combined treatment with crizotinib. In June 2018, based on the in vitro results and the molecular analysis, he started a combined treatment of pembrolizumab (3 cycles of 200 mg every 3 weeks) and crizotinib (250 mg 2 times a day every day) treatment. Remarkably, four weeks into treatment, the patient showed an excellent clinical and RECIST response without any adverse events. PET-CT at two months post-treatment initiation showed near CR and no evidence of any harmful metabolic activity (Figure 2E). He was continuously treated for the next two months until he was suddenly admitted to the hospital with severe pneumonia. The patient had a rapid and dramatic clinical deterioration with respiratory failure which resulted in his death. A summary of the patient diagnostics, treatment duration, and responsiveness to PD-1 treatment are shown in figure 1. Representative PET-CT scans of disease progression and treatment response are presented in Figure 2A-G.
Methods
Genetic studies
Genetic studies were performed using the FoundationOne CDx test (Foundation Medicine, Inc.). Microsatellite instability and tumor mutational burden (TMB) were evaluated using targeted next-generation sequencing. Quantitative proteomics analysis was performed using the GPS Cancer test from NantHealth company (NantHealth, Inc.).
Tumor ex vivo screening analysis (TEVA)
TEVA was done to evaluate the efficacy of agents on the tumor cells. Two small molecules, crizotinib (5uM; MedChem Express, HY-50878) and afatinib (2uM; MedChem Express, HY-10261), were used according to a previously published protocol [52]. Immunohistochemical staining was performed on tissue sections for Ki67 (Vector laboratories), p-ERK (Cell signaling Technology), and TUNEL (TREVIGEN) according to the manufacturer's protocol. Images were taken by a panoramic scanner (3D Histech), analyzed by HistoQuantTM software (3D Histech).
CD8+ T cell activation assays
CD8+ T cells were collected and isolated from the patient by standard methodology. Isolated CD8+ T cells were then analyzed by IFN-γ ELISA and CD107a degranulation assays, as previously reported [52]. For IFN-γ ELISA and CD107a degranulation assay, xenograft derived cells at 0.5×105 cells/ well concentration were seeded in 96 well U-bottom plates and incubated with and without crizotinib (5uM) overnight. Following incubation, 2×105 CD8 T cells/ well were added with fresh RPMI containing 20U IL-2 and mixed with the tumor cells. Pembrolizumab at a concentration of 10ug/mL was then added to respective wells. For IFN-γ ELISA after overnight incubation, the supernatant was collected from the wells and assayed by a standard IFN-γ ELISA assay (ELISA MAX, Biolegend). For CD107a degranulation assay, following incubation, 1×105 of total cells were resuspended in 200 ul of fresh complete RPMI and subjected to Cytospin™ 4 Cytocentrifuge (Thermo Fischer Scientific).
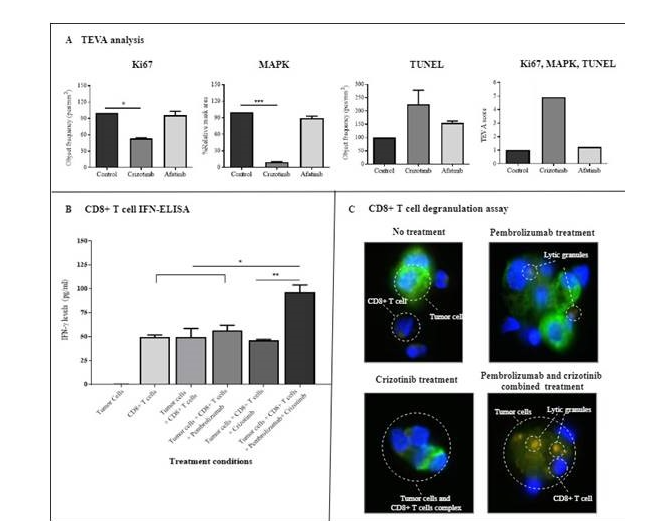
Figure 3:In vitro treatment efficiency studies. The patient's tumor cells were isolated from pembrolizumab resistant tumor specimens. Autologous CD8+ T cells were isolated from the patient peripheral blood during his nonresponsive pembrolizumab stage. Panel A. The patients' Tumor Ex Vivo Analysis (TEVA), which measured the tumor cell proliferation (Ki67), activation (mitogen-activated protein kinase (MAPK) signaling), and apoptosis (terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)). Based on the patient molecular analysis (Fig. 1), crizotinib and afatinib (as control) were evaluated for their treatment efficiency. Crizotinib had a significant anti-tumor effect on tumor cell proliferation and activation. Analysis of variance (ANOVA) comparing responses to control are marked with * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Panel B shows patient in vitro CD8+ T cell activation results. The patient's tumor cells were incubated with autologous CD8+T cells alone, with pembrolizumab only, with crizotinib only or with both drugs. IFN-γ secretion levels, as a marker for CD8+ T cell response, were measured. As indicated, a combined crizotinib and pembrolizumab treatment showed a significant positive effect on patient CD8+ T cell capacity to regain sensitivity to pembrolizumab. Statistical significance was determined by Analysis of variance (ANOVA) comparing all treatment responses. Statistical analysis: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Panel C shows the treatment effect of crizotinib on CD8+ T cell activation against the patient's pembrolizumab resistant tumor cells by immunofluorescent microscopy. As shown, single-drug treatment by pembrolizumab or crizotinib had no effect in comparison to combined crizotinib + pembrolizumab that induced tumor lysis. Index color is as follows; light blue staining represents tumor cells, CD8+ T cell nuclei (by DAPI, dark blue ), tumor cell cytokeratin 14 marker stained in green, and CD107a representing lytic granulose stained in orange.
Slides were fixed in 4% paraformaldehyde for 10 mins, permeabilized with 0.1% Triton X-100 in PBS for 5 mins, blocked with 5% FBS and 0.01% Triton X-100 in PBS for 30 mins. Afterward, slides were incubated overnight with APC-conjugated human CD107a mAb (BioLegend) and Anti-Cytokeratin 14 antibody (Abcam). Following incubation, slides were washed and incubated with goat anti-rabbit Alexa-Fluor 488 (Abcam) for 90 mins. Slides were rewashed and incubated with DAPI (Biolegend). Finally, slides were mounted into VectaMount® AQ Aqueous Mounting Medium (Vector Laboratories, H-5501), and images were taken by a panoramic scanner (3D Histech).
Statistical Analysis
Graphical and statistical analyses were performed using GraphPad Prism 7.0 software. Statistical analysis was performed using the one-way ANOVA and Bonferroni multiple comparison test.
Discussion
PD-1/PD-L1 blockade has become a standard treatment for patients with advanced NSCLC [1]. However, most patients will develop primary resistance or secondary resistance. There is no effective treatment option for these patients who show resistance. The mechanisms of PD-1 blockade resistance are involved with tumor intrinsic and external factors [22–24]. These included low infiltrating T lymphocytes, poor specificity of antigen-specific cytotoxic T cells, and inadequate T cells response to PD-1/PD-L1 axis signaling. The tumor microenvironment was also shown to play a pivotal role in resistance affecting the immunogenicity of tumor cells, the sensitivity of tumor cells to cytotoxic T cells, and impairing presenting function of antigen-presenting cells [7].
Recent evidence has pointed at specific driver oncogenes involved in PD-1/PD-L1 blockade treatment efficacy [25], and suggested that genetic heterogeneity may provide a vital substrate for the emergence of resistance during treatment-selective pressures [26]. Such changes, which could also alter the tumor microenvironment, are gaining higher recognition of their importance [27]. Out of those genetic heterogeneity changes, MET overexpression has been shown to have a pivotal role in cancer progression, proliferation, and survival [28]. Furthermore, it has been recently suggested to play an essential part in resistance to anti-PD-1/PD-L1 immunotherapies [28,29]. There is evidence that MET overexpression is associated with the expression of immunoregulatory molecules such as PD-L1 [30–32] and Indoleamine-2,3-dioxygenase (IDO) in cancer cells [33]. Furthermore, MET may also affect inflammation-induced PD-L1 expression. IFN-γ induced upregulation of PD-L1 expression represents a negative control mechanism to limit the T-cell responses' magnitude [34,35]. The IFN-γ pathway may also be modulated by MET inhibition. In clinical practice, PD-1/PD-L1 therapy was shown to be less effective in tumors with driver mutations, among them MET [36]. However, the importance of MET protein expression and gene amplification has been questioned, as clinical studies using MET targeting therapies in patients with protein overexpression or gene amplification have produced disappointing results [37]. Nevertheless, MET presents an attractive therapeutic target as there are clinically available inhibitors that may modulate cancer cells' ability to evade anti-tumor immune responses [15,19,37].
Although we still have a long way to fully understand the best treatment in patients with PD-1 blockade resistance, this case report, and additional evidence from our other similarly treated patients (data not shown), indicates that acquired resistance to PD-1/PD-L1 treatment was observed upon de novo MET overexpression. To examine MET overexpression's potential role as a mediator of PD-1/PD-L1 acquired resistance, we evaluated potential treatment strategies using the patient tumor and CD8+ T cells. We assessed the effect of crizotinib and pembrolizumab as a single and combined therapy strategy. Treatment efficiency was determined based on proliferation, associated signaling pathway, apoptosis, and IFN secretion (for T cell activation). Our "in vitro" results showed that crizotinib significantly reduced tumor cell proliferation and the MAPK signaling pathway and induced a higher level of apoptosis. The combined treatment of crizotinib and pembrolizumab significantly induced the patient’s CD8+ T cell IFN-γ secretion levels, which was not observed after either a single crizotinib pembrolizumab treatment.
Crizotinib's positive impact on T cell activation is in line with recent data, suggesting that crizotinib can improve treatment outcomes via off-target effects [38]. Early reports from a phase I clinical study suggest patients with high MET amplification may respond better to treatment with crizotinib [39]. Moreover, treatment response with crizotinib in patients with advanced NSCLC with MET amplification showed a success rate of 33% [40]. Crizotinib increases immunogenic cell death by T-lymphocyte infiltration and, thus, improves treatment outcomes [41]. Tyrosine kinase inhibitor (TKI) drugs such as crizotinib were shown to modulate the expression of inhibitory receptors on T cells, or interfere with T cell function or differentiation, both synergizing with ICI therapies [16,42]. In our study, crizotinib treatment primarily affects cell proliferation and signal transduction rather than cell apoptosis. Therefore, we hypothesized that the MET overexpression blockade helps convert an unresponsive microenvironment to a responsive one via CD8+ T cell activation. This T cell activation could have been mediated through signaling pathways within or outside the PD-1 axis [16].
ICI-mediated antitumor responses rely on the expression of PD-L1 in tumors and the infiltration of T cells into the tumor microenvironment (TME) [43]. The absence of T cells in the TME can lead to resistance to immunotherapy and so-called immune-desert tumor phenotypes [44]. These tumors may lack T-cell priming due to the absence of tumor antigens, defective antigen processing, and presentation machinery or impaired DC-T-cell interactions [45]. There is growing evidence that the activation of specific oncogenic pathways, like MET, is related to such “cold tumor” phenotype and the potential for immunotherapy resistance. Thus, crizotinib treatment against MET could help convert a “cold tumor” into a responsive one (“hot tumor”). Further work is still needed to support this hypothesis and identify the specific action mechanism [46].
At present, combination therapy strategies are being developed to improve PD-1/PD-L1 blockade efficacy in various tumor types, including NSCLC. These include combinations with radiation therapy, chemotherapy, and small molecular inhibitors, including TKI [47,48]. In this regard, the potential effectiveness of TKI and anti-PD-1/PD-L1 therapy combinations have recently been described [49]. It has been shown that such a combination approach improved the median overall survival of advanced NSCLC patients [50,51]. TKI combined treatment was suggested to "prime" the immune system toward a superior ICI-effective response. However, a significant amount of pre-clinical investigation is required before TKI, and anti-PD-1/PD-L1 combined therapy, can be considered. This combination has caused a relatively high incidence of treatment-related adverse events, with toxicity being the most important limitation [52,53]. For example, in the CheckMate 370 trial, NSCLC patients treated with nivolumab plus crizotinib developed severe hepatic toxicities, leading to discontinuing the treatment combination [54]. These toxicities, including fatal side effects, also tended to appear earlier and rapidly in the course of the combination treatment [40,47]. Caution is needed in patients receiving combination therapy based on PD-1/PD-L1 inhibitors and TKI. Further clinical studies are required to define the safety profile and potential risk in such patients before introducing this therapy into routine clinical practice.
Clinical oncologists are still unclear about which treatment regimen should be selected after PD-1 blockade failure. Here, we report a patient with advanced NSCLC with acquired PD-1 blockade resistance, which showed a rapid resensitization to PD-1 blockade therapy following combined treatment of crizotinib against a de novo MET overexpression. The mechanism underlying this is still unclear and needs to be further explored. Furthermore, since crizotinib was not used as a sole treatment, it remains unknown whether the positive clinical effect seen in our patient could have been achieved using crizotinib alone as a single treatment course. However, the excellent clinical and RECIST response observed points to a link between the de novo MET overexpression and PD-1 acquired resistance. This finding suggests that targeted therapy against MET could have a vital role in reversing PD-1 blockade resistance in a patient with MET overexpression.
Conclusions
It remains challenging to clarify the resistance mechanisms of immunotherapy since they are complex and dynamic. Further understanding of acquired resistance mechanisms of immunotherapy will help clinicians make reasonable combination treatment decisions to increase survival and avoid additional toxicity for lung cancer patients. Combination therapy based on PD-1/PD-L1 inhibitors may provide a promising therapeutic strategy for resistant PD-1 pathway blockades in NSCLC. Comprehensive genotyping of targetable driver oncogenes such as MET should be performed, along with the determination of PD-L1 status, to optimize treatment stratification. The evidence thus far indicates that the role of aberrant MET activity on the expression of immunoregulatory pathways and PD-L1 blockage resistance by tumor cells, is worthy of further investigation in NSCLC, as is the potential combination of MET targeting therapies with ICI.
Data Availability Statement
The datasets generated and method used in this study are available on supplemental source file and upon request to the corresponding author.
Ethics Statement
This study involving a human participant was reviewed and approved by the ethics committee of Soroka Medical Center. The patient provided written, informed consent to participate in the ex-vivo biomarker studies. All biopsies and whole blood samples were taken under appropriate informed consent approved by the institutional review board of Soroka Medical Center. Written informed consent was obtained from the patients or next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
The study was designed by NP, LR, WK, ME, and AP. In vitro and in vivo works were performed by KK and SG. Data acquisition was made by NP, SG did LR, SG, KK, YAC, and AS. Data analysis and interpretation, KK, LR, NP, WS, SG, KK, YAC, ME, AP and AS. Manuscript preparation was made by AS. Manuscript editing was done by LR, JC, WK, ME, AP, and NP.
Funding
This work was supported by the Israel Science Foundation grant 1188/16 (to A. Porgador), the United State-Israel Binational Science Foundation grant 2015344 (to A. Porgador), the Israeli Ministry of Science and Technology/German Cancer Research Center program Grant CA172 (to A. Porgador), the Israeli Ministry of Science and Technology grant 54180 (to A. Porgador). This work was also funded by the Israeli Cancer Research Foundation (ICRF, 17-1693-RCDA) (to M. Elkabets), Israel Science Foundation grant 700/16 (to M. Elkabets), United State—Israel Binational Science Foundation grant 2017323 (to M. Elkabets). An Alon Fellowship supports M. Elkabets for outstanding young researchers.
Conflict of Interest
N. Peled is the advisor for and receives personal honoraria from Astra-Zeneca, Boehringer Ingelheim, Bayer, BMS, Lilly, MSD, Novartis, NovellusDx, Pfizer, Roche, Takeda, Foundation Medicine, Gaurdant360; and participates as an investigator in clinical trials with Astra-Zeneca, Boehringer Ingelheim, Bayer, BMS, MSD, Novartis, Roche, Takeda.
References
1. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018; 62:29–39. doi:10.1016/j.intimp.2018.06.001
2. Planchard D, Popat S, Kerr K, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;doi:10.1093/annonc/mdy474
3. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018; 378:2078–2092. doi:10.1056/NEJMoa1801005
4. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (80- ). 2015;348:124–128. doi:10.1126/science.aaa1348
5. Zimmermann S, Peters S, Owinokoko T, Gadgeel SM. Immune Checkpoint Inhibitors in the Management of Lung Cancer. Am Soc Clin Oncol Educ B. 2018; 682–695. doi:10.1200/EDBK_201319
6. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018; 118:9–16. doi:10.1038/bjc.2017.434
7. Shergold AL, Millar R, Nibbs RJB. Understanding and overcoming the resistance of cancer to PD-1/PD-L1 blockade. Pharmacol Res. 2019; 145: doi:10.1016/j.phrs.2019.104258
8. Xia L, Liu Y, Wang Y. PD‐1/PD‐L1 Blockade Therapy in Advanced Non‐Small‐Cell Lung Cancer: Current Status and Future Directions. Oncologist. 2019; 24: doi:10.1634/theoncologist.2019-io-s1-s05
9. Kong X, Lu P, Liu C, et al. A combination of PD‑1/PD‑L1 inhibitors: The prospect of overcoming the weakness of tumor immunotherapy (Review). Mol Med Rep. 2021; 23:1–16. doi:10.3892/mmr.2021.12001
10. Wang F, Wang S, Zhou Q. The Resistance Mechanisms of Lung Cancer Immunotherapy. Front Oncol. 2020; 10: doi:10.3389/fonc.2020.568059
11. Sanchez-Cespedes M. B2M, JAK2 and MET in the genetic landscape of immunotolerance in lung cancer. Oncotarget.2018; 9:35603–35604. doi:10.18632/oncotarget.26277
12. Saigi M, Alburquerque-Bejar JJ, Mc Leer-Florin A, et al. MET-Oncogenic and JAK2-Inactivating Alterations Are Independent Factors That Affect Regulation of PD-L1 Expression in Lung Cancer. Clin Cancer Res. 2018; 24:4579–4587. doi:10.1158/1078-0432.CCR-18-0267
13. Miranda O, Farooqui M, Siegfried JM. Status of Agents Targeting the HGF/c-Met Axis in Lung Cancer. Cancers (Basel). 2018; 10: doi:10.3390/cancers10090280
14. Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011; 3:S7–S19. doi:10.1177/1758834011422556
15. Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. 2006; 12:3657–3660. doi:10.1158/1078-0432.CCR-06-0818
16.Sun X, Li C-W, Wang W-J, et al. Inhibition of c-MET upregulates PD-L1 expression in lung adenocarcinoma. Am J Cancer Res. 2020; 10:564–571.
17. Robinson KW, Sandler AB. The Role of MET Receptor Tyrosine Kinase in Non‐Small Cell Lung Cancer and Clinical Development of Targeted Anti‐MET Agents. Oncologist (2013) 18:115–122. doi:10.1634/theoncologist.2012-0262
18. Drilon A, Cappuzzo F, Ou SHI, et al. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J Thorac Oncol. 2017; 12:15–26. doi:10.1016/j.jtho.2016.10.014
19. Kim ES, Salgia R. MET pathway as a therapeutic target. J Thorac Oncol. 2009; 4:444–447. doi:10.1097/JTO.0b013e31819d6f91
20. Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol 2013. 31:1089–1096. doi:10.1200/JCO.2012.43.9422
21. Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003; 4:915–925. doi:10.1038/nrm1261
22. Lei Q, Wang D, Sun K, et al. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front Cell Dev Biol. 2020; 8:672. doi:10.3389/fcell.2020.00672
23. Boyero L, Sánchez-Gastaldo A, Alonso M, et al. Primary and acquired resistance to immunotherapy in lung cancer: Unveiling the mechanisms underlying of immune checkpoint blockade therapy. Cancers (Basel). 2020; 12:1–36. doi:10.3390/cancers12123729
24. Yuan Y, Adam A, Zhao C, et al. Recent Advancements in the Mechanisms Underlying Resistance to PD-1/PD-L1 Blockade Immunotherapy. Cancers (Basel). 2021; 13:663. doi:10.3390/cancers13040663
25. Aspeslagh S, Chabanon RM, Champiat S, et al. Understanding genetic determinants of resistance to immune checkpoint blockers. Semin Cancer Biol. 2020. doi:10.1016/j.semcancer.2019.12.020
26. Riaz N, Havel JJ, Makarov V, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017; 171:934-949 e16. doi:10.1016/j.cell.2017.09.028
27. Pogrebniak KL, Curtis C. Harnessing Tumor Evolution to Circumvent Resistance. Trends Genet. 2018; 34:639–651. doi:10.1016/j.tig.2018.05.007
28. Papaccio F, Della Corte CM, et al. HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. Int J Mol Sci. 2018; 19: doi:10.3390/ijms19113595
29. Sabari JK, Leonardi GC, Shu CA, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018; 29:2085–2091. doi:10.1093/annonc/mdy334
30. Yang H, Chen H, Luo S, et al. The correlation between programmed death-ligand 1 expression and driver gene mutations in NSCLC. Oncotarget. 2017; 8:23517–23528. doi:10.18632/oncotarget.15627
31. Albitar M, Sudarsanam S, Ma W, et al. Correlation of MET gene amplification and TP53 mutation with PD-L1 expression in non-small cell lung cancer. Oncotarget (2018) 9:13682–13693. doi:10.18632/oncotarget.24455
32. Titmarsh HF, O’Connor R, Dhaliwal K, Akram AR. The Emerging Role of the c-MET-HGF Axis in Non-small Lung Cancer Tumor Immunology and Immunotherapy. Front Oncol (2020) 10:54. doi:10.3389/fonc.2020.00054
33. Wang D, Saga Y, Sato N, et al. The hepatocyte growth factor antagonist NK4 inhibits indoleamine-2, 3-dioxygenase expression via the c-Met-phosphatidylinositol 3-kinase-AKT signaling pathway. Int J Oncol. 2016; 48:2303–2309. doi:10.3892/ijo.2016.3486
34. Blank C, Brown I, Peterson AC, et al. PD-L1/B7H-1 Inhibits the Effector Phase of Tumor Rejection by T Cell Receptor (TCR) Transgenic CD8+ T Cells. Cancer Res. 2004; 64:1140–1145. doi:10.1158/0008-5472.CAN-03-3259
35. Garcia-Diaz A, Shin DS, Moreno BH, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017; 19:1189–1201. doi:10.1016/j.celrep.2017.04.031
36. Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019; 30:1321–1328. doi:10.1093/annonc/mdz167
37. Titmarsh HF, O’Connor R, Dhaliwal K, Akram AR. The Emerging Role of the c-MET-HGF Axis in Non-small Cell Lung Cancer Tumor Immunology and Immunotherapy. Front Oncol. 2020; 10:54. doi:10.3389/fonc.2020.00054
38. Titmarsh HF, O’Connor R, Dhaliwal K, et al. The Emerging Role of the c-MET-HGF Axis in Non-small Lung Cancer Tumor Immunology and Immunotherapy. Front Oncol. 2020; 10:54. doi:10.3389/fonc.2020.00054
39. Camidge DR, Otterson GA, Clark JW, et al. Crizotinib in patients (pts) with MET-amplified non-small cell lung cancer (NSCLC): Updated safety and efficacy findings from a phase 1 trial. J Clin Oncol. 2018;36:9062–9062. doi:10.1200/jco.2018.36.15_suppl.9062
40. Camidge DR, Ou S-HI, Shapiro G, et al. Efficacy and safety of crizotinib in patients with advanced c-MET -amplified non-small cell lung cancer (NSCLC). . J Clin Oncol. 2014;32:8001–8001. doi:10.1200/jco.2014.32.15_suppl.8001
41. Liu P, Zhao L, Pol J, et al. Author Correction: Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun. 2019; 10:1883. doi:10.1038/s41467-019-09838-y
42. Huemer F, Leisch M, Geisberger R, et al. Combination Strategies for Immune-Checkpoint Blockade and Response Prediction by Artificial Intelligence. Int J Mol Sci. 2020; 21:2856. doi:10.3390/ijms21082856
43. Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020; 20:662–680. doi:10.1038/s41568-020-0285-7
44. Hegde PS, Chen DS. Top 10 Challenges in Cancer Immunotherapy. Immunity. 2020; 52:17–35. doi:10.1016/j.immuni.2019.12.011
45. Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. 2021; 11:5265–5286. doi:10.7150/thno.58390
46. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017; 541:321–330. doi:10.1038/nature21349
47. Boutros C, Tarhini A, Routier E, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016; 13:473–486. doi:10.1038/nrclinonc.2016.58
48. Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017; 8: doi:10.3389/fphar.2017.00561
49. García-Aranda M, Redondo M. Targeting protein kinases to enhance the response to anti-PD-1/PD-l1 immunotherapy. Int J Mol Sci. 2019; 20: doi:10.3390/ijms20092296
50. Molife C, Hess LM, Cui ZL, Li XI, Beyrer J, Mahoui M, Oton AB. Sequential therapy with ramucirumab and/or checkpoint inhibitors for non-small-cell lung cancer in routine practice. Futur Oncol. 2019; 15:2915–2931. doi:10.2217/fon-2018-0876
51. Felip E, de Braud FG, Maur M, et al. Ceritinib plus Nivolumab in Patients with Advanced ALK-Rearranged Non–Small Cell Lung Cancer: Results of an Open-Label, Multicenter, Phase 1B Study. J Thorac Oncol. 2020;15:392–403. doi:10.1016/j.jtho.2019.10.006
52. Lin JJ, Chin E, Yeap BY, et al. Increased Hepatotoxicity Associated with Sequential Immune Checkpoint Inhibitor and Crizotinib Therapy in Patients with Non–Small Cell Lung Cancer. J Thorac Oncol. 2019;14:135–140. doi:10.1016/j.jtho.2018.09.001
53. Oshima Y, Tanimoto T, Yuji K, et al. EGFR-TKI-associated interstitial pneumonitis in nivolumab-treated patients with non-small cell lung cancer. JAMA Oncol. 2018; 4:1112–1115. doi:10.1001/jamaoncol.2017.4526
54. Spigel DR, Reynolds C, Waterhouse D, et al. Phase 1/2 Study of the Safety and Tolerability of Nivolumab Plus Crizotinib for the First-Line Treatment of Anaplastic Lymphoma Kinase Translocation — Positive Advanced Non–Small Cell Lung Cancer (CheckMate 370). J Thorac Oncol. 2018; 13:682–688. doi:10.1016/j.jtho.2018.02.022
Received: January 19, 2022;
Accepted: February 14, 2022;
Published: February 16, 2022.
To cite this article : Roisman LC, Peled N, Kundu K, et al. Crizotinib Restored Pembrolizumab Response in Advanced Non- Small Cell Lung Cancer. European Journal of Respiratory Medicine. 2022; 4(1): 268-275. doi: 10.31488/EJRM.126.
© 2022 Roisman LC, et al.